Le soufre
Le soufre est un élément essentiel pour tous les êtres vivants. Il intervient notamment dans la
formule de deux acides aminés, la cystéine et la méthionine et, par conséquent, dans de nombreuses
protéines. L’essentiel du soufre sur Terre se trouve dans les sédiments
et dans les roches sous forme de minéraux sulfatés (gypse, CaSO4) et sulfurés (pyrite, FeS2), et au
niveau des océans, sous forme de sulfates SO4 2-, qui constituent un des réservoirs les plus significatifs
de soufre pour la biosphère. Les sédiments marins sont riches en composés soufrés plus ou moins réduits. Il existe en effet de nombreux niveaux d’oxydation du soufre.
Le sulfate est la forme la plus oxydée (degré d’oxydation du soufre +VI) et l’un des principaux anions
présents dans l’eau de mer avec des concentrations voisines de 28 mM. Le sulfate va servir
d’accepteur d’électrons pour les bactéries sulfato-réductrices (Jørgensen and Bak, 1991). Les sulfures
sont la forme la plus réduite du soufre (degré d’oxydation du soufre -II). Le soufre élémentaire S0
(degré d’oxydation du soufre 0) est surtout connu sous la forme de cristaux de couleur jaune. On peut
le trouver sous forme native notamment dans les régions volcaniques. Il existe d’autres composés
soufrés correspondant à différents degrés d’oxydation du soufre, comme par exemple le thiosulfate
(S2O3 2-, degré d’oxydation du soufre +II) et le sulfite (SO3 2-, +IV). Cette large gamme d’états
d’oxydation du soufre le place au centre d’un large éventail de réactions d’oxydo-réduction.
Un des processus chimiques les plus importants au niveau des sédiments marins riches en
matière organique est la décomposition de cette dernière par l’intermédiaire de la sulfato-réduction
bactérienne. La réduction des sulfates et du thiosulfate présents dans l’eau de mer nécessite des donneurs d’électrons organiques comme le lactate et le pyruvate, les hydrocarbures, les acides gras volatils ou encore les sucres (ou de l’hydrogène moléculaire, produit de la fermentation de la matière organique, principal donneur d’électrons pour les bactéries sulfato-réductrices). Cette réduction ne se produit donc que si d’importantes quantités de matières organiques sont présentes. Dans la plupart des sédiments marins, le taux de réduction des sulfates est donc limité par l’apport de carbone. Plusieurs composés soufrés inorganiques sont d’importants accepteurs d’électrons pour la respiration anaérobie.
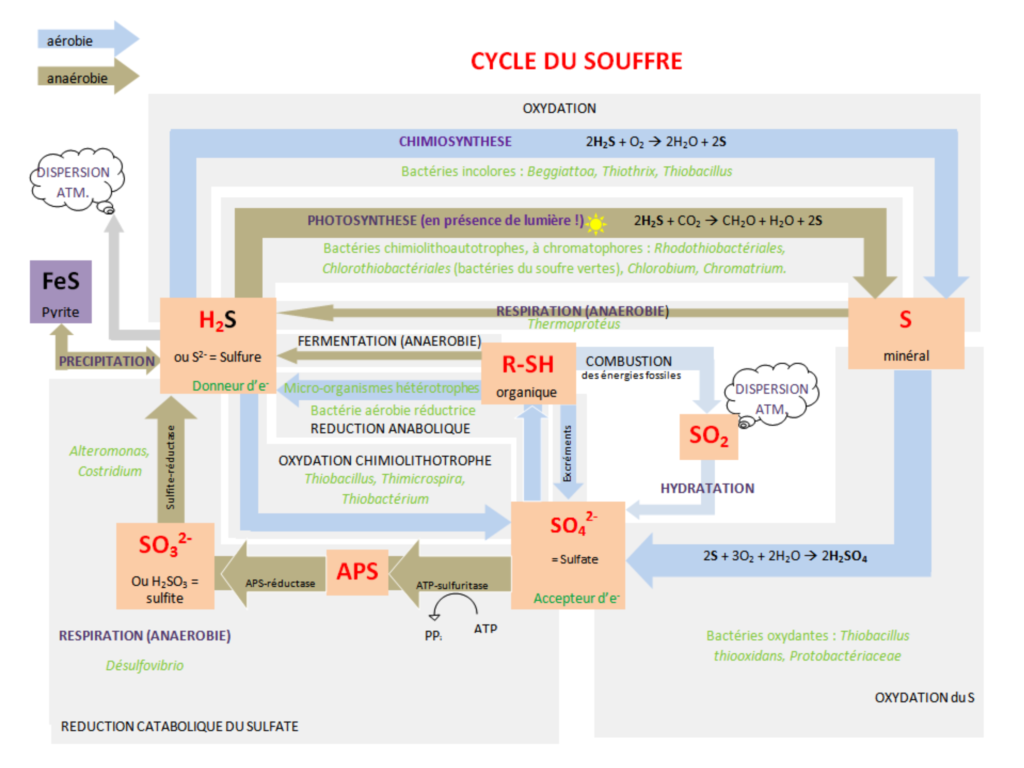
Le produit final est l’hydrogène sulfuré H2S. La réduction du sulfate et des formes oxydées intermédiaires du soufre par les bactéries peut servir à la synthèse de la biomasse. L’hydrogène sulfuré formé sera alors immédiatement converti en soufre organique sous forme d’acides aminés, on parle de réduction assimilative.
Dans le cas de la réduction dissimilative en revanche, l’H2S est excrété, permettant ainsi la minéralisation de la matière organique. Le sulfate est beaucoup moins favorable en tant qu’accepteur d’électrons que l’oxygène ou les nitrates. Une des enzymes clefs de la sulfato-réduction est la sulfite reductase, qui catalyse la réduction du sulfite (SO3 2-) en sulfures (H2S). La sulfite reductase est présente dans tous les groupes majeurs connus de sulfato-réducteurs. A l’exception d’Archaeoglobus, un membre des Archaea, tous les procaryotes sulfato-réducteurs connus
appartiennent au domaine des Bacteria. Certaines de ces bactéries seraient d’ailleurs les partenaires syntrophiques des archées réalisant la méthanogenèse et l’oxydation anaérobie du méthane. L’oxydation de l’H2S peut aller jusqu’à
la production de sulfate, en passant par divers intermédiaires dont le soufre élémentaire
En effet, le sulfure peut être séquestré dans des hydrates de gaz ou se complexer avec le fer sous forme de monosulfide de fer ou de pyrite (espèce minérale composée de disulfure de fer, FeS2) dans des sédiments riches en fer. Par ailleurs, en conditions oxygénées, l’ion sulfure HS va être spontanément et rapidement oxydé à pH neutre. Pour ces raisons, l’oxydation microbienne significative des sulfures, principalement aérobie, ne se produira qu’au niveau des zones où l’H2S qui provient des zones anoxiques, en quantité suffisante, rencontre l’O2 des zones oxygénées, c’est-à-dire les zones de transition oxique/anoxique. Les procaryotes oxydant les sulfures peuvent former des
biofilms à la surface des sédiments, aussi appelés tapis microbiens, ou être associés de façon symbiotique avec des invertébrés tels que les vésicomyidés.
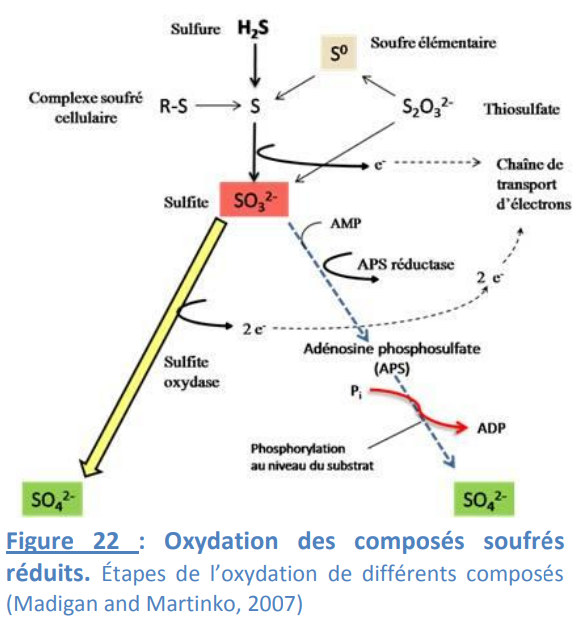
Le soufre élémentaire S0 ou encore le thiosulfate S2O3 2-, qui ont des degrés d’oxydation
intermédiaires par rapport au sulfure et au sulfate, peuvent également être aisément oxydés ou réduits
par des microorganismes. Les bactéries qui oxydent le soufre élémentaire sont souvent fortement attachées aux cristaux de soufre. Ces espèces intermédiaires peuvent aussi être l’objet de disproportionation, c’est-à-dire
séparation d’un composé en deux nouveaux composés, l’un étant plus oxydé et l’autre plus réduit que
le substrat d’origine, ou de comproportionation, c’est-à-dire une réaction dans laquelle deux composés
qui comprennent un même élément mais à des degrés d’oxydation différents vont réagir ensemble
pour former un produit qui contient le même élément à un degré d’oxydation intermédiaire entre les
deux substrats. Le soufre élémentaire (degré d’oxydation du soufre 0) pourra ainsi être disproportioné
en sulfate (+VI) et en sulfure (-II). C’est l’hypothèse avancée plus haut au sujet du métabolisme des
sulfato-réducteurs associés aux ANMEs réalisant l’AOM. De la même façon, le sulfure d’hydrogène et le dioxyde de soufre pourront par exemple être comproportionés en soufre élémentaire.
Le sulfure d’hydrogène est le résultat de la combinaison de deux éléments : l’hydrogène H et le soufre S, soit la formule H2S. Le sulfure d’hydrogène est un hydrure à l’odeur désagréable d’œuf pourri. Appelé également : hydrogène sulfuré, mais aussi quelquefois hydrure de soufre, monosulfure de dihydrogène ou sulfure de dihydrogène. C’est un gaz acide qui réagit avec les solutions aqueuses basiques et les métaux tels que l’argent. C’est la raison pour laquelle les bijoux argentés noircissent lorsqu’ils sont longuement exposés à l’atmosphère polluée. Le sulfure d’argent résultant de la réaction est de couleur noire. Le sulfure d’hydrogène est produit par la dégradation des protéines contenant du soufre et est responsable d’une grande partie de l’odeur fétide des excréments et des flatulences. Il peut résulter de décomposition bactérienne de la matière organique. Il est également produit par les déchets humains et animaux. Il peut donc s’accumuler dans les réseaux d’assainissement et corroder les tuyaux qu’ils soient en béton ou en métal. Il peut faire suffoquer les égoutiers. Le sulfure d’hydrogène est naturellement présent dans le pétrole, le gaz naturel, les gaz volcaniques et les sources chaudes. Le sulfure d’hydrogène peut également provenir des activités industrielles, telles que la transformation des produits alimentaires, du traitement des eaux usées, des haut-fourneaux, des papeteries, des tanneries, des raffineries de pétrole ou d’algues vertes en décomposition comme sur les plages de la baie de Lannion. La plus grosse partie du sulfure d’hydrogène dans l’atmosphère provient de sources naturelles : eaux sulfurées, marais salins et zones d’activité géothermique, volcanisme. Le niveau ambiant pour le sulfure d’hydrogène est estimé à 0,3 µg/m³. Dans les environs de la ville de Rotorua (Nouvelle Zélande), ville proche de zones à forte activité géothermique. Des concentrations bien plus importantes ont même été mesurées dans les environs de papeteries (plusieurs centaines de µg/m³).
Comme le sulfure d’hydrogène pose plus de problèmes de nuisances olfactives que de réels problèmes de santé publique, il est intéressant d’envisager le nombre de dépassements du seuil olfactif. Dans la littérature scientifique, les valeurs citées pour le seuil de perception du sulfure d’hydrogène varient fortement suivant les sources. Aussi, mon choix s’est porté sur la valeur de 7 µg/m³ sur ½ heure recommandée par l’OMS., en gardant à l’esprit que certaines tranches de la population pourront percevoir l’H2S à des teneurs plus faibles, alors que d’autres ne sentiront rien ! Inflammable, la substance se décompose en brûlant, et en en présence d’un excès d’oxygène, la combustion est totale : 2 H2S + 3 O2 >>> 2 SO2 + 2 H2O (production de dioxyde de soufre SO2, très toxique). Très toxique par inhalation, aux conditions « normales » de pression, c’est un gaz plus lourd que l’air (1,45 kg m3-1 à 15°C). Le sulfure d’hydrogène (H2S) est un gaz dangereux. On le classe parmi les gaz asphyxiants chimiques parce qu’il entre immédiatement en réaction chimique avec l’hémoglobine du sang, ce qui empêche le transport de l’oxygène jusqu’aux tissus et aux organes vitaux du corps. Il est produit par la décomposition anaérobie de matières organiques comme le fumier. À faible concentration, c’est un gaz facile à détecter du fait de son odeur caractéristique d’œuf pourri, mais à des concentrations élevées, il provoque la paralysie du nerf olfactif et donc la perte d’odorat. Une personne exposée au sulfure d’hydrogène peut donc avoir une fausse impression de sécurité. À forte concentration, le sulfure d’hydrogène cause instantanément la paralysie et la mort. Le soufre, élément indispensable à la vie, n’est présent chez les êtres vivants qu’à raison de 1 à 2 % de leur masse. Il l’est sous forme d’acides aminés soufrés (cystéine, méthionine) ainsi que dans les centres fer-soufre des protéines. Dans la nature, le soufre est le plus souvent sous forme de sulfate, dans la mer et la roche, ou de sulfure de sels métalliques (pyrite par exemple). Il peut également être présent sous forme élémentaire, c’est le soufre que l’on rencontre près des volcans. Le cycle du soufre fait intervenir dans la nature un certain nombre de microorganismes : les sulfatoréducteurs anaérobies tels que Desulfovibrio ou Clostridium ; Les bactéries phototrophes (Chromatium, Thiopedia…), des bactéries aérobies telles que Thiobacillus ou Achromatium, des archaebactéries comme les bactéries méthanogènes.

Cas des tapis microbiens
Le terme de « tapis microbien » vient de la structure macroscopique que ces écosystèmes
microbiens adoptent. Ils forment en effet des voiles denses à la surface des sédiments. Les
microorganismes filamenteux à la taille imposante qui les forment se développent en une sorte de
masse enchevêtrée avec les particules de sédiments. Les tapis microbiens se développent souvent dans des environnements caractérisés par des conditions de vie extrêmes qui excluent les métazoaires. Ces derniers dévoreraient dans le cas contraire les microorganismes et les empêcheraient de former un tapis. Les microorganismes formant les tapis microbiens sont généralement autotrophes et puisent leur énergie dans l’oxydation de composés
réduits comme le méthane ou le sulfure. Ce sont des bactéries géantes filamenteuses affiliées à l’ordre des Thiotrichales, principalement des genres Beggiatoa, Thiomargarita et Thiothrix, qui forment souvent ces tapis microbiens.
Ces bactéries géantes peuvent être blanches, jaunes ou oranges et former d’immenses tapis microbiens
de plusieurs mètres de diamètre, qui recouvrent la surface des fonds marins au niveau d’écosystèmes
riches en sulfures, comme c’est le cas des suintements froids et dans les écosystèmes hydrothermaux
sédimentaires. La coloration blanche est due à la nature réfringente des granules de soufre élémentaire qu’elles peuvent stocker dans leur cytoplasme. Les tapis orange sont quant à eux souvent associés aux zones riches
en hydrocarbures, et il a été proposé que la pigmentation des bactéries qui les composent soit reliée à leur source principale de carbone. Cependant, une étude récente a par ailleurs proposé l’existence d’un lien entre la couleur des filaments et leur affiliation phylogénétique. Les Thiotrichales, des genres Beggiatoa et Thiothrix sont des bactéries autotrophes sulfo-oxydantes. Elles sont localisées aux interfaces oxiques/anoxiques en surface des sédiments, ce qui leur permet d’utiliser l’oxygène de l’eau de mer pour oxyder les composés soufrés. Ces bactéries sont capables de se mouvoir par glissement et de s’enfouir dans les sédiments lorsque les conditions environnementales sont moins favorables. Ces tapis microbiens sont souvent utilisés comme indicateurs visuels de sorties de fluides au niveau des écosystèmes chimiosynthétiques marins.
Selon Influence des communautés microbiennes sédimentaires sur la répartition faunistique dans les sites hydrothermaux et les zones d’émissions de fluides froids du bassin de Guaymas par Perrine Cruaud UBO 2014
L’azote et le phosphore
Les rejets d’azote et de phosphore proviennent de l’agriculture et des eaux usées domestiques. Ils affectent l’équilibre des écosystèmes aquatiques en accélérant notamment le phénomène d’eutrophisation voire une dystrophie dans certain cas. Cette contamination oblige les collectivités locales qui utilisent cette ressource pour leur alimentation en eau potable à procéder à un traitement plus complet, qui se traduit par des coûts plus élevés.
Les matières azotées et phosphatées, notamment l’ammoniaque, sont produites par les villes et par quelques industries. Selon leur forme, ces matières ont des effets différents :
L’azote organique, comme toutes substances organiques, contribue à la désoxygénation de l’eau.
L’azote ammoniacal est gênant pour la fabrication d’eau potable et génère un poison, le gaz ammoniac, dangereux pour le poisson.
L’azote nitrique (celui des nitrates) amène une surproduction d’algues avec des inconvénients écologiques et esthétiques très graves. Sa présence, en grande quantité dans l’eau potable, est contre-indiquée, surtout pour les nourrissons (la norme maximale en nitrates de l’eau potable est 50 mg/l).
L’amélioration de la qualité générale de l’eau observée ces dernières années alliée à une forte charge en azote mais surtout en phosphore, favorise le développement de végétaux. Ces proliférations végétales sont à l’origine de nombreuses nuisances, dont les plus fréquentes sont une coloration des eaux, la présence d’odeurs, une gêne à l’écoulement des eaux, et surtout des mortalités massives de poissons par asphyxie.
L’objet est de valoriser les données acquises sur les réseaux « physico-chimie » en prenant en compte à la fois des facteurs à l’origine de l’eutrophisation (phosphore et azote) et les conséquences de ces phénomènes : la chlorophylle a est un indicateur de la quantité du phytoplancton et oxygène dissout dont la sursaturation indique des développements excessifs de végétaux aquatiques de différente nature. La valeur du rapport azote sur phosphore (N/P) permet de situer les linéaires des cours d’eau dont les conditions nutritives favorisent l’apparition d’algues du groupe des cyanobactéries fixatrices d’azote atmosphérique. Ces algues constituent des fleurs d’eau ou blooms en cas de prolifération excessive, et sont à l’origine des plus graves nuisances pouvant aller jusqu’à la mort d’organismes par intoxication.
Quatre niveaux d’eutrophisation des cours d’eau sont décrits à partir des valeurs moyennes annuelles des paramètres N et P et des valeurs maximales des paramètres O2.
Eléments limitants P/N Rapport N/P dans la cellule algale: N/P » 7-10 (en masse). Si N/P>10 le phosphore est le facteur limitant, si N/P <7, l’azote est le facteur limitant. Dans ces milieux, le phosphore est souvent l’élément qui limite la prolifération végétale. Les algues ont besoin entres autres pour leur croissance de carbone, d’azote et de phosphore. Elles ont besoin de ces trois éléments selon le ratio C:N:P de 106:16:1 (exprimé en nombres d’atomes). Dans les eaux naturelles, le ratio entre les quantités d’N et de P disponibles est généralement supérieur à 16. Par conséquent, même en cas de pollution du milieu par l’azote, si aucun apport de P n’est réalisé, les algues ne pourront pas se développer. La pollution des eaux par le phosphore est donc très souvent à l’origine de développements algaux importants et conduit à la dystrophisation des milieux.
En ce qui concerne la toxicité directe du nitrate sur les mammifères, seules des concentrations de plusieurs dizaines de grammes par litres sont susceptibles de causer des problèmes, ce qui, fort heureusement n’intervient qu’extrêmement rarement en milieu naturel. En réalité, la toxicité directe de très fortes doses de nitrate sur les organismes n’est jamais observée dans la nature. Les problèmes liés à la consommation des nitrates sont le plus souvent indirects et découlent de sa transformation en nitrite. Le risque lié au nitrite tient au fait qu’il constitue un oxydant puissant. Chez l’homme, l’absorption chronique de doses excédant quelques dizaines de mg/L peut conduire à une cyanose (méthémoglobinémie) chez les nourrissons et certaines personnes âgées. La détoxication intervient par sécrétion des excédents (reins, glandes salivaires). Bien qu’aucun phénomène de cancérogenèse n’ait jamais été observé avec ces deux types de sels, certains composés (nitrosamines) présents dans l’alimentation ou dont la formation est induite par le nitrite de façon endogène sont toutefois suspectés. Chez les organismes aquatiques, les méthémoglobinémies sont aussi étudiées chez les poissons, notamment le saumon atlantique. En milieu aquatique, la présence de nitrate s’accompagne invariablement de celle de nitrite et d’ammonium. Le niveau d’oxygénation ainsi que la demande biologique et chimique en oxygène conditionnent en effet les équilibres entre ces trois espèces. Selon le cas, le nitrite peut connaître des teneurs toxiques pour les animaux. Le seuil généralement admis est de 0,5 mg/L de nitrite (10,9 µM de N-NO2). Le risque lié au nitrate est donc fonction des conditions du système considéré, que cela soit sur la vasière ou dans les claires ostréicoles. Ces dernières sont toutefois plus exposées dans la mesure où elles présentent un certain confinement et parfois des charges organiques importantes. Les connaissances acquises aujourd’hui permettent de mieux cerner les déterminismes de ces crises, voire d’orienter la production microalgale.
La présence de l’azote ammoniacal dans l’eau à des concentrations élevées a un impact sur le milieu aquatique. Ses effets dépendent de la température et du pH de l’eau. En effet, pour un pH de 7.4 à 8.5, la vie aquatique est affectée par des concentrations d’environ 2 mg/L, alors que pour une température de 15°C, la teneur en ammoniac dangereuse est de l’ordre de 6 mg/L à pH de 7. Les nitrates et les nitrites constituent la forme la plus abondante d’azote dans l’eau. Bien que naturellement présents en faibles quantités dans les eaux de surface, des concentrations trop élevées de nitrites et de nitrates peuvent être toxiques pour la faune aquatique et provoquer une maladie infantile (méthémoglobinémie). (GLS mémotec24). La responsabilité des nitrates dans les cancers digestifs fait débat. Mais il est démontré que les nitrates transformés en nitrosamines sont cancérigènes chez de nombreux animaux. (Libération, 9 mars 2005)
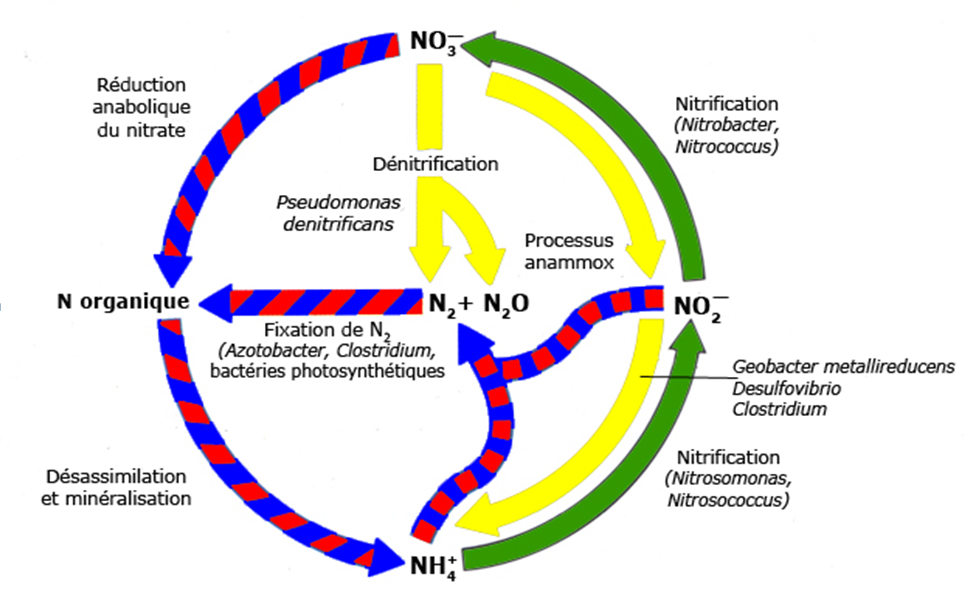
L’azote inorganique possède plusieurs états d’oxydation allant de –3 (ammonium et groupement amine) à +5 (nitrate). Les nombreuses transformations de l’azote permettent à ces 26 éléments de circuler entre le compartiment atmosphérique et les milieux terrestres et aquatiques et de déterminer en partie la productivité biologique des habitats et l’épuration des milieux riches en azote. Fixation de l’azote. L’azote atmosphérique n’est pas disponible pour la plupart des organismes en raison de l’énergie nécessaire pour casser la triple liaison dans N2, ce qui rend ce composé quasiment inerte. Tout l’azote disponible pour le biota dérive initialement de la fixation de cet azote, soit par la foudre, soit par quelques espèces de microbes spécialisés. La fraction issue des éclairs est minime. La fixation biologique d’azote se produit soit à partir de microbes qui vivent dans les sols, les sédiments ou les eaux, soit en association symbiotique avec les racines de plantes terrestres ou aquatiques. Le processus de fixation d’azote se produit surtout dans les habitats pauvres en azote. Le taux de fixation d’azote est généralement contrôlé par le rapport N:P. L’ajout de P stimule la fixation d’azote. Suivant le milieu considéré, la fixation de l’azote peut être conduite par des bactéries et des archae qui convertissent l’azote moléculaire en ammoniac. Les cyanobactéries sont les principaux microorganismes fixateurs d’azote dans les milieux aquatiques. Ces microorganismes tirent l’énergie nécessaire à la fixation de l’azote grâce à la photosynthèse. L’activité de l’Homme a très largement impacté le cycle global de l’azote, puisqu’en plus des processus naturels de fixation d’azote, l’homme produit des engrais azotés à partir de la réaction Haber-Bosch. Les volumes fabriqués de nos jours par ce processus sont à peu près équivalents à la quantité d’azote fixé naturellement. Assimilation de l’azote La disponibilité de NH4 + et NO3 – est variable selon les environnements. Les producteurs primaires peuvent assimiler les deux formes. L’azote inorganique est ensuite converti en groupe amine (-NH2). Le DON peut également être une source d’azote dans des milieux appauvris en NH4 + et NO3- . Seules les molécules organiques de faibles poids moléculaires comme les urées et les acides aminés peuvent être rapidement disponibles. Ammonification ou minéralisation La décomposition de l’azote de la matière organique débute par la libération d’acides aminés et autres composés organiques azotés simples qui peuvent être directement assimilés. La transformation de ces composés en ammonium est appelée ammonification. En fonction de la complexité structurale de la matière organique, l’ammonification peut être une simple réaction de désamination ou une série de processus métaboliques complexes faisant intervenir des enzymes. La nitrification est un processus aérobie en deux étapes au cours desquelles l’ammoniac ou les ions ammonium sont oxydés en nitrite puis en nitrate. Ce processus est couplé à la fixation de carbone par des bactéries chimioautotrophes, généralement classées dans les genres Nitrosomonas et Nitrobacter. La première étape de la nitrification (nitrosation) est réalisée par les bactéries nitrosantes, parfois nommées AOB (Ammonia–Oxidizing Bacteria), qui sont strictement aérobies. Ces bactéries oxydent l’ammonium en nitrite. La seconde étape de la nitrification est réalisée par les bactéries nitratantes, qui consomment immédiatement le nitrite. Ce groupe bactérien convertit le nitrite en nitrate grâce à une enzyme, la nitrite oxydoréductase. Dénitrification La dénitrification est un processus hétérotrophe anaérobie où le nitrate sert d’accepteur d’électron pour l’oxydation du carbone organique. Le nitrate est converti en azote gazeux (N2O et N2). Le processus de dénitrification est contrôlé par le passage de conditions aérobies à des conditions anaérobies. La dénitrification peut toutefois se produire en présence d’oxygène. Les bactéries dénitrifiantes convertissent le nitrate en oxyde nitrique (NO) via le nitrite, puis en oxyde nitreux (N2O) et enfin en azote gazeux N2. La dénitrification se fait essentiellement dans les sédiments et les zones ripariennes. En milieu aquatique, elle est réalisée quand le milieu devient anoxique. Volatilisation de l’ammoniac Ce processus physico-chimique est lié à l’équilibre entre la forme dissoute et gazeuse de NH3. L’ammoniac peut être volatilisé des eaux vers l’atmosphère dans les zones humides naturelles ou artificielles. Ce processus devient significatif uniquement lorsque le rapport entre NH3 et NH4 + devient important. Ceci se produit dans les eaux dont le pH est basique. En effet, le rapport NH3 :NH4 + est de 0,1 à pH 8,3 ; il est de 1 à pH 9,3. De tels pH sont cependant courants en journée dans les milieux aquatiques peu profonds, durant les périodes où l’activité photosynthétique des macrophytes fait considérablement baisser la pression partielle de CO2 dissous, et donc augmenter le pH. Autres processus de transformation des nitrates Les principaux processus d’extraction des nitrates des milieux aquatiques sont ceux décrits ci-dessus (assimilation et dénitrification). Depuis 20 ans, de nombreux travaux ont mis en évidence des processus microbiens alternatifs, parmi lesquels la réduction dissimilatrice du nitrate en ammonium, la dénitrification via l’oxydation des sulfures ou du fer ferreux et l’anammox. L’oxydation de l’ammonium en N2, avec le nitrite comme accepteur d’électron est appelée anammox. Ce processus est important dans les zones très productives telles que les upwellings. Il a été mis en évidence dans tout type de milieu pauvre en oxygène où l’ammonium est oxydé en nitrite (sédiments, zones humides…). Cycle biogéochimique de l’azote :
- Le phosphore
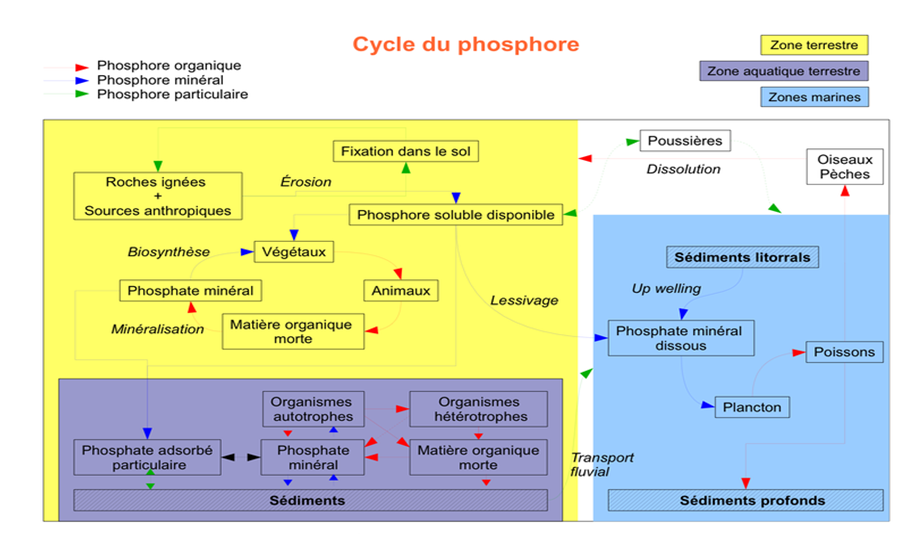
Un nutriment limitant pour la productivité biologique en eau douce. Le phosphore est considéré comme le nutriment le plus limitant dans la plupart des écosystèmes d’eau douce. A l’échelle des temps géologiques, sa disponibilité est considérée comme étant le contrôle majeur de la concentration en oxygène de l’atmosphère. Le phosphore est utilisé en tant qu’engrais en agriculture. Le phosphore primaire a une origine géologique. Dans l’eau, le phosphore est essentiellement sous forme d’ion phosphate correspondant à l’équilibre de dissociation de l’acide orthophosphorique (H3PO4 ↔ H2PO4 – + H+ ↔ HPO4 2- + 2H+ ↔ PO4 3- + 3H+). Toutefois, en raison de la forte affinité de ces anions pour les surfaces des particules, les phosphates sont peu solubles. Le phosphore dissous existe aussi sous des formes organiques plus ou moins facilement décomposables. La minéralisation de la litière des sols via les différents métabolismes hétérotrophiques, les activités agricoles et les décharges d’eaux usées sur le bassin versant libèrent les ions phosphate dans le milieu, où ils deviennent disponibles pour la production biologique. Les teneurs en phosphore dans les eaux lacustres dépendent également des sources internes comme le recyclage du phosphore dans les sédiments ou le recyclage de la biomasse dans la colonne d’eau. Sous des conditions limitées en phosphore, le phosphore organique dissous (DOP) peut servir de source indirecte en phosphore pour les micro-organismes sous l’action de phosphatases qui permettent l’hydrolyse du DOP ou par minéralisation bactérienne. Le DOP peut être libéré lors de la lyse de la matière organique, excrété par les organismes de la colonne d’eau ou bien apporté par le bassin versant. Cette forme de phosphore peut constituer une source importante de phosphore pour le développement de la biomasse. L’adsorption des ions phosphates sur les oxydes et oxyhydroxydes de fer et d’aluminium a été très abondamment étudiée en sciences du sol, parce que (1) le phosphore est un nutriment limitant dans les écosystèmes terrestres et (2) le piégeage par adsorption du phosphore naturel ou issus des engrais impacte les niveaux de production agricole et forestière. Les oxydes/oxyhydroxydes de fer(III) (Fe(III)-ox) peuvent être dissous par réduction ou bien former des sulfures de fer dans les milieux réducteurs, ce qui libère le phosphore associé. En conséquence, les conditions rédox du milieu jouent un rôle important dans la biodisponibilité du phosphore. Le principal puits de phosphore en milieu aquatique est l’enfouissement dans les sédiments. Les échanges entre les sédiments et l’eau surnageante se font au travers des processus biogéochimiques benthiques, parmi lesquels la minéralisation du P organique, le cycle rédox du fer et les flux benthiques de phosphore. Le phosphore associé à la matière organique qui arrive au fond est donc rapidement minéralisé en phosphate dissous par les organismes benthiques. Il peut alors diffuser dans les eaux interstitielles pour regagner la colonne d’eau. Par conséquent, seule une partie du phosphore qui atteint le sédiment y est enfoui. Une portion du phosphore des eaux interstitielles issu de la minéralisation de la matière organique peut être adsorbée sur les Fe(III)-ox détritiques ou authigéniques dans la zone oxydée des sédiments. Ainsi, la séquestration du phosphore dépend largement de la capacité d’adsorption du sédiment. Cette capacité est directement liée à la quantité d’oxydes de fer présents dans les sédiments. Une fois que ces particules se retrouvent dans la zone anoxique des sédiments, soit par enfouissement soit par de l’advection par bioturbation, la fraction la plus réductible des Fe(III)-ox peut être réduite et dissoute, ce qui libère les ions phosphate. Une partie des phosphates libérés dans la zone anoxique peut précipiter sous des formes authigènes d’apatite lorsque les eaux sont fortement sur saturée. Le phosphore peut ainsi être enfoui pour de longues périodes sous la forme de P-organique, d’oxydes de fer réfractaires et d’apatite. D’autres formes de minéraux phosphatés authigéniques ont été décrites dans des sédiments marins ou lacustres. Par exemple, la vivianite, un phosphate de fer ferreux, se forme dans les sédiments anoxiques où le fer dissous peut s’accumuler dans les eaux interstitielles. Ainsi, pour pouvoir décrire le comportement du phosphore dans les environnements aquatiques et les sols, il est nécessaire de connaître la spéciation solide de cet élément. Le P faiblement adsorbé est la fraction qui peut être facilement désorbé dans l’eau quand la concentration en phosphate dissous diminue, ou quand la salinité augmente, comme dans les estuaires.
Le phosphore total (Ptot) est la somme du phosphore dissous et en suspension. Les formes du phosphore se présentent sous les formes suivantes : Minéral dissout, on trouve les orthophosphates et les polyphosphates. Organique dissout, les POD.
Minéral particulaire, les apatites, phosphore ferrique et phosphore absorbé par les MES. Organique particulaire, les matériaux cellulaires comme l’ADN, l’ARN ou l’ATP. Les bactéries et les algues utilisent les POD grâce aux enzymes, phosphatases alcaline ou acide, en fonction du pH, leur efficacité varie. Les bactéries mortes ou vivantes rejettent du POD particulaire ou minéral, toutes les formes sont recyclables. Il existe deux flux croisés de phosphate entre l’eau et les sédiments : Précipitation en condition aérobie des apatites, sel ferrique… Solubilisation en condition anaérobie, réduction des phosphates en phosphites (P(RO)3) et en phosphines ((PH3)n )(GLS mémotec23).
Ce sont des micronutriments essentiels pour les organismes aquatiques, et se retrouvent naturellement dans l’eau à de faibles concentrations. Ils proviennent du lessivage du sol et du substrat rocheux. Les ions métalliques se trouvent en milieu aqueux sous diverses formes: libres, complexés avec des ligands simples organiques ou minéraux inorganiques, complexés avec des ligands macromoléculaires ou colloïdaux, adsorbés ou incorporés aux particules en suspension organiques ou inorganiques et adsorbés ou assimilés par les organismes. Les formes chimiques prises par les métaux sont importantes non seulement pour comprendre leurs toxicité et biodisponibilité, mais aussi, pour conceptualiser et évaluer les types de traitement des eaux. Leur importance a été clairement démontrée dans les traitements basés sur la coagulation, floculation ou adsorption utilisées, par exemple, pour éliminer les phosphates, fluorures ou la matière organique. D’autre part, les procédés utilisés pour le traitement des pollutions métalliques présentent généralement des rendements médiocres.