Les divers facteurs de pollution physico-chimique posent un problème difficile et urgent, menaçant la qualité des eaux superficielles, mais aussi les nappes souterraines et les eaux marines. La pollution par les sels minéraux est fréquente provient des effluents domestiques, des engrais agricoles et de certains établissements industriels et sont retrouvés dans les nappes souterraines. Les dérivés du phosphore sont issus des lessives et participent, avec ceux de l’azote, aux phénomènes d’eutrophisation. Les métaux représentent une forme particulière de pollution chimique. Ils sont déversés par les industries ou par lessivage des sols urbains sur lesquels se sont déposés les diverses pollutions liées au trafic, au chauffage…, et peuvent s’accumuler dans les rivières, ainsi qu’en certains points du littoral, et ainsi atteindre la flore et la faune aquatique. Cette forme de pollution a été et est encore à l’origine de foyers d’intoxications. Même à faible dose par le consommateur, ils peuvent s’accumuler et être responsables de pathologies à long terme affectant, selon les cas, le système nerveux, le rein et le squelette. La pollution organique par les détergents non biodégradables, les hydrocarbures, les solvants, est une autre forme de pollution chimique. Elle est inquiétante par son aspect ubiquitaire, persistant, insidieux et polyvalent et l’on se pose de plus en plus la question d’éventuels effets perturbateurs endocriniens ; certaines de ces substances pourraient en effet interférer avec le système hormonal et influencer négativement les processus de synthèse, de sécrétion, de transport, l’action ou l’élimination des hormones, comme cela a été démontré sur diverses espèces car ce n’est pas la dose qui fait le poison.
De même les stations d’épurations rejettent des eaux qui répondent à des critères déterminés par des lois administratives, mais les milieux de rejets répondent à des lois naturelles.
Aujourd’hui, des réglementations existent à l’échelle mondiale comme européenne. En 2001, la Convention de Stockholm a été adoptée ; en 2014, 152 pays ont approuvé cette convention visant à diminuer voire stoppé la production de polluants organiques persistants. Sur le plan européen, depuis 1976, la directive européenne 76/464/CEE, abrogée en 2000 par la Directive 2000/60/CE ou Directive Cadre sur l’Eau (DCE), vise à réduire les rejets de substances dangereuses dans le milieu aquatique (Directive n°76/464/CEE, 1976, Directive n°2000/60/CE, 2000). La DCE a pour objectif de définir l’état écologique et chimique de chaque cours d’eau européenne et d’en restaurer le « bon état ».
L’évaluation de l’exposition requiert de déterminer la présence de contaminants dans les écosystèmes en estimant leur concentration et leur biodisponibilité. Cette partie fait appel à la chimie analytique environnementale. De par les différentes techniques de séparation et les détecteurs aujourd’hui disponibles, les instruments analytiques permettent d’analyser une large gamme de polluants organiques dans l’environnement. Les analyses ciblées sur une pré- sélection de molécules sont privilégiées pour la surveillance du milieu. Ces analyses étant optimisées sur des composés prédéfinis, elles permettent d’atteindre des niveaux de concentrations pouvant être qualifiés de traces. Cependant, elles ne prennent pas en compte toutes les molécules présentes dans les échantillons et sont susceptibles de sous-estimer un danger potentiel. Pour pallier ces limites, de nouvelles techniques analytiques de recherches de molécules ont été développées comme la spectrométrie de masse haute résolution (HRSM). Néanmoins afin d’appréhender un risque réel, il est nécessaire de caractériser des effets écologiques et par conséquent de faire appel au domaine de la biologie. Dans l’évaluation du risque, cette étape est la caractérisation des effets. L’approche se base sur des essais multi-espèces pouvant être effectués en microcosmes et mésocosmes afin d’essayer de déterminer les effets de mélange de contaminants et la possible bioaccumulation le long des chaines trophiques. L’évaluation des effets combinés, dit « effet cocktail » est l’une des principales difficultés à résoudre et doit absolument être prise en compte pour une évaluation correcte et pertinente du risque.
Le milieu estuarien
Lieux de rencontre entre les eaux continentales et les eaux marines, les estuaires sont définis comme des zones de transition. Les interactions entre les paramètres physicochimiques propres à chacune des masses d’eau engendrent la formation d’écosystèmes complexes et fluctuants. En effet, les marées, ajoutées aux débits ainsi qu’aux courants de circulation, ont pour effet majeur d’homogénéiser les masses d’eaux continentale et marine. Les estuaires comme des environnements dynamiques où les paramètres physico-chimiques ainsi que biologiques présentent une variabilité spatiale et temporelle élevée. En effet, le mélange des deux masses d’eau induit de forts gradients des paramètres environnementaux (tels que la salinité et les matières en suspension MES) au sein d’un continuum fluvioestuarien. Les estuaires sont également considérés comme des exutoires des bassins versants et a fortiori des réceptacles d’apports amont riches en sels nutritifs et matières organiques. Ils sont donc des zones de transit de matière et sont dès lors des environnements privilégiés pour sa transformation (floculation, adsorption…) et sa dégradation (consommation, activités microbiennes). La diversité des apports en matière organique (MO) au sein des estuaires complexifie également la compréhension du fonctionnement de ces systèmes. Elle peut être allochtone, c’est-à-dire produite en dehors du système estuarien (d’origine strictement fluviale ou océanique, anthropique…), ou autochtone, issue d’une production strictement estuarienne (phytoplancton, microphytobenthos, macrophytes…). De la même façon que les paramètres physicochimiques, la contribution de ces sources au pool de matière organique en suspension ou sédimentaire (organique et inorganique) dépend de la localisation au sein du continuum fluvioestuarien et également de la période de l’année. La variabilité de la composition du pool de matière organique est donc variable spatialement et temporellement à une échelle tidale (coefficients, débits), saisonnière (crues et étiages) et interannuelle. Certains estuaires, notamment les estuaires macrotidaux, présentent un phénomène naturel relativement complexe ayant de nombreuses conséquences sur le fonctionnement de l’estuaire. Ce phénomène résulte de la circulation résiduelle engendrée par l’écoulement différentiel des eaux douces et des eaux salées, ainsi que de l’asymétrie de l’onde de marée. En lien avec l’écoulement des eaux douces en surface (moins denses) et des eaux salées en 4 profondeur, l’accumulation élevée de MES est à l’origine de la formation d’une zone de turbidité maximum : le « bouchon vaseux ». Le BV est issu du piégeage de particules fines du fait d’une circulation résiduelle en amont de l’intrusion saline. Il est soumis à des migrations longitudinales et à des cycles de dépôt et de remise en suspension à différentes échelles de temps selon l’intensité des débits fluviaux. C’est donc cette variabilité naturelle rapide des paramètres physico-chimiques qui rend la compréhension et la dynamique d’un estuaire si complexes. L’importance accrue des impacts anthropiques, en particulier durant les vingt dernières années, a par ailleurs amplifié cette complexité. En effet, le développement des activités humaines a fait perdre aux plus grands estuaires certaines de leurs fonctionnalités écologiques (fragmentation des habitats, modification de la biodiversité et de l’hydrodynamique…), allant même jusqu’à menacer leur dynamique. Ne représentant que 6% de la superficie des eaux marines, les écosystèmes côtiers et littoraux concentrent pas moins de 60% de la population mondiale et bientôt 75% aux alentours de 2025. En terme financier, les estuaires participent à 33% de la valeur économique des systèmes marins. En plus d’être hautement productifs, ils possèdent un rôle écologique fondamental au regard de leur importance en tant que zone de nourricerie, de croissance et de couloir migratoire pour un grand nombre d’espèces de poissons et d’oiseaux. Depuis toujours, l’homme associe les écosystèmes côtiers à de très forts enjeux économiques et sociétaux. Les estuaires accueillent des activités industrielles (raffineries, usines) avec en parallèle les infrastructures nécessaires à leur bon fonctionnement (tunnels, ponts…). Les activités portuaires se sont fortement développées à partir du milieu du XIXème siècle, favorisant les échanges économiques mais modifiant de façon importante la morphologie des estuaires. Les estuaires sont également un service majeur lorsque l’on parle d’alimentation avec les activités de pêche, d’aquaculture et d’agriculture. De plus, leur eau est utilisée en tant que ressource de choix pour l’industrie et l’agriculture (refroidissement des centrales nucléaires, irrigation…). Les estuaires ont un rôle majeur aussi en tant que services culturels, patrimoniaux et touristiques. Dans ce contexte actuel de changement global, de nombreux travaux ont mis en avant que les principales évolutions actuelles (hors modifications aigues telles que des pollutions/aménagements ponctuels) sont l’augmentation de la température et le phénomène de « marinisation » (intrusion saline accrue). Ces modifications ont un effet sur les aires de répartition des espèces, sur la phénologie et la diversité des organismes, ainsi que sur leur réactivité, c’est-à- dire leur temps d’adaptation vis-à-vis de ces modifications environnementales Cet ensemble de changements observés à l’échelle des communautés engendre majoritairement un déclin, voire la disparition, de certaines espèces ou, à l’inverse, l’apparition d’autres espèces. Certaines espèces invasives et/ou opportunistes peuvent bénéficier de ces nouvelles conditions et s’installer dans ces systèmes.
Les systèmes estuariens sont caractérisés par de profonds changements dans les propriétés chimiques de l’eau et souvent par de hautes activités biologiques. Ces variations affectent significativement la distribution des éléments et leurs transferts vers la zone côtière adjacente. C’est particulièrement vrai dans le cas des métaux, des nutriments et de la matière organique. A chaque flux de la marée, la résistivité de l’eau diminue avec la concentration en sel qui augmente, ce qui provoque un relargage de métaux des complexes organométalliques. Au reflux de la marée les complexes perdent leurs ions métalliques, ce qui empêche l’évolution des degrés d’oxydation naturel pour atténuer leur toxicité. (Selon : Transfert de métaux entre eau et suspensions dans les estuaires Khaled Sioud) Evaluation de l’état écologique des masses d’eau de transition dans le cadre de la DCE Etude de la pertinence du suivi des peuplements du microphytobenthos estuarien. L’évaluation de l’état écologique des eaux de transition repose sur les poissons, les invertébrés benthiques, les macroalgues et angiospermes et le phytoplancton. Cet EQB, qui est pertinent pour les eaux côtières, est beaucoup plus compliqué à interpréter pour les eaux de transition. L’alternative envisagée est de considérer les microalgues qui se développent à marée basse et forment des biofilms, parfois de taille kilométrique, à la surface des sédiments. Les eaux de transition et les eaux côtières ne bénéficient pas d’un tel historique de travaux de recherche.
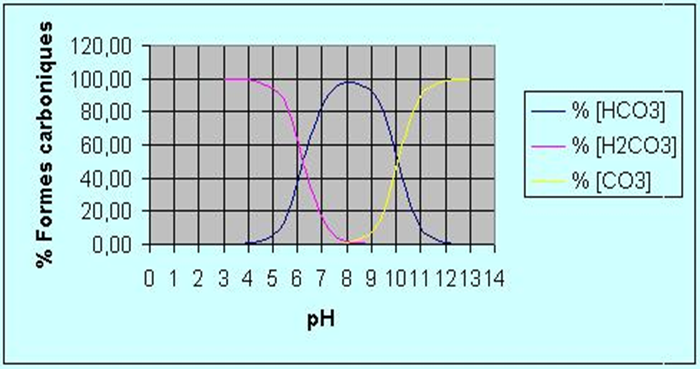
Le pH
Le pH est une mesure de l’harmonie des concentrations entre les ions H3O+ et OH– de l’eau H2O (autoprotolyse de l’eau : H2O ↔ (H3O+ + OH–) s’équilibre en fonction de l’ionisation de l’eau). Les eaux naturelles ont un pH voisin de 7, le plus souvent compris entre 6 et 8. Plus le pH est bas, plus la solution est dite acide. Le pH influe sur la croissance animale et végétale sur l’assimilation des oligoéléments par les animaux et les végétaux sur l’activité enzymatique bactérienne et sur la complexation.
Plusieurs espèces de poissons et autres organismes aquatiques ne peuvent supporter une eau trop acide. La pollution atmosphérique et les précipitations acides demeurent la plus importante source d’acidité des plans d’eau. Le pH subit des fluctuations journalières en fonction des variations de la concentration en CO2, liées à l’activité photosynthétique (la nuit l’eau est plus acide que le jour). Si le pH est supérieur à 7, l’eau est propice à la végétation. Si par contre il est inférieur à 7, l’eau est peu propice à la vie et rende les poissons plus sensibles aux parasites.
(Pluies acides)
Potentiel d’oxydoréduction ou redox
Permet de qualifier une eau résiduaire et de la classer en milieu plutôt oxydant ou plutôt réducteur. Une oxydation signifie qu’un ion ou une molécule perd des électrons, mais cette réaction s’accompagne automatiquement d’un gain d’électrons par un autre réactif et ce dernier processus est appelé réduction : Dans les eaux résiduaires, l’oxygène est rapidement consommé par oxydation hétérotrophe de la matière organique. Lorsque l’on passe en milieu réducteur, la tendance est à la fermentation des matières organiques, c’est-à-dire un processus de putréfaction qui implique des bactéries anaérobies. L’interconversion chimique de l’oxygène en eau est également importante car elle contrôle la valeur effective du potentiel redox. Le logarithme de la pression partielle en oxygène rH est un indice, analogue au pH, qui fournit une mesure quantitative du potentiel oxydant ou réducteur du milieu. Le milieu est considéré comme réducteur lorsque rH < 27 et oxydant pour. Par ailleurs, un test de putrescibilité peut aisément être réalisé. Il consiste à mesurer le temps nécessaire pour décolorer une certaine quantité de bleu de méthylène ajoutée à l’eau résiduaire. La décoloration indique que l’on passe en milieu réducteur, avec tendance à la putréfaction des matières organiques. Le potentiel redox est une mesure des conditions d’aération des milieux aqueux. Un potentiel redox positif signifie de bonnes conditions d’aération avec une concentration importante d’oxygène dissous (milieu aérobie); par contre un potentiel redox négatif est observé lors d’une diminution de l’oxygène dissous (milieux anoxiques). La diminution de l’oxygène dissous peut être provoquée par la dégradation de la matière organique, surtout dans les conditions d’eutrophisation, c’est à dire, de croissance anormalement élevée des plantes grâce à la présence de nitrates et phosphates qui favorisent ce développement. La diminution de l’oxygène dissous peut avoir un effet sur les organismes aquatiques comme les poissons, mais aussi sur la mobilisation des métaux. Comme nous l’avons indiqué les hydroxydes et oxydes de fer et de manganèse adsorbent une quantité importante des métaux lourds, mais ces substrats sont susceptibles de se dissoudre avec la variation des conditions d’oxygénation et d’entraîner la libération des métaux associés. Les oxydes de manganèse précipitent à la surface des sédiments seulement à potentiel redox très positif, c’est à dire dans des bonnes conditions d’aération, mais se dissolvent en Mn2 + lorsque le potentiel redox devient très négatif. Les hydroxydes ferriques (Fe III), se dissolvent plus facilement que les oxydes de manganèse.
L’équilibre calcocarbonique
L’étude des caractéristiques physico-chimiques des eaux, ainsi que leur action sur le milieu environnant, sont souvent traitées comme des cas particuliers à cause de la complexité des relations entre les ions présents.
Ces équilibres sont instables en raison des variations de la température et des teneurs en gaz dissous (évasion ou dissolution), ce qui amène de nouvelles réactions et conséquences sur le milieu en contact avec l’eau.
En particulier, les problèmes de traitement, d’agressivité, d’incrustation ou de corrosion sont générés par les variations d’équilibres physico-chimiques et posent des problèmes techniques pour le captage, l’adduction et la distribution des eaux. L’étude de ces problèmes nous amène presque toujours à ce que l’on peut appeler :
Mettre une eau à l’équilibre, c’est lui donner, par un traitement approprié, des caractéristiques stables dans le temps qui s’écoule entre son stockage après traitement et son utilisation par le consommateur. L’équilibre de l’eau dépend de deux facteurs :
* ses caractéristiques propres,
* la nature des matériaux susceptibles de se trouver à son contact.
La recherche de cet équilibre nécessite la mise en œuvre de deux moyens d’action :
* opérer un choix judicieux de ces matériaux qui devront être les plus inertes possible à l’action de l’eau pour que l’équilibre de celle-ci ne soit pas compromis,
* donner à l’eau des caractéristiques intrinsèques stables, compte tenu de ces matériaux.
Il va de soi que s’il s’agit d’installations anciennes, l’équilibre de l’eau ne peut être obtenu que dans l’amélioration des caractéristiques de celle-ci et non dans le choix des matériaux qui n’a d’objet que s’il s’agit d’installations nouvelles. Les inconvénients consécutifs à un défaut d’équilibre des eaux sont dus à :
* leur agressivité vis-à-vis des calcaires, bétons et ciments,
* leur corrosivité vis-à-vis des métaux,
* leur caractère incrustant.
Dans les deux premiers cas, les ouvrages et équipements concernés sont endommagés et même détruits et l’eau acquiert turbidité et coloration. Dans le dernier cas, les canalisations sont rétrécies, parfois même obstruées et ne transitent plus les débits prévus.
- Equilibre ionique des eaux
* Gaz (ou anhydride carbonique) – CO2, se combinant partiellement à l’eau, il forme l’acide carbonique H2CO3 qui est l’agent essentiel de l’agressivité de l’eau dans laquelle il est partiellement dissocié en ions.
* Oxygène, O2 – Azote, N2 (se dissolvent dans l’eau sans se dissocier)
- Hydrogène sulfuré (H2S et lien interne sur le gaz), légère dissociation : H2S <<< >>> 2 H+ + S2-
* Sels de bases fortes et d’acides forts : sels totalement dissociés en ions. Les plus important sont les sulfates SO42- et les chlorures Cl–. Une eau qui a été par exemple en contact avec des terrains gypseux (sulfate de calcium – CaSO4) se chargera d’anions SO42- et de cations Ca2+, selon la réaction : Ca SO4 <<< >>> Ca2+ + SO42-
* Sels de bases fortes et d’acides faibles
sels presque totalement dissociés en ions. Les plus importants sont les sels de calcium, Ca2+, et en particulier les sels de l’acide carbonique : bicarbonate de calcium, Ca(HCO3)2 et carbonate de calcium CaCO3.
La turbidité
La turbidité est une façon de mesurer la transparence de l’eau. Elle s’exprime en NTU (unités de turbidité par néphélémétrie) et est causée par la présence de matières en suspension (MS), telles que l’argile, le limon, les particules organiques, le plancton et les autres organismes microscopiques. Une turbidité trop élevée empêche la pénétration de la lumière dans la colonne d’eau et peut ainsi diminuer la croissance des algues et des plantes aquatiques. Elle peut aussi protéger les bactéries et les virus contre les procédés de désinfection de l’eau potable, car la plus grande partie se trouve sous formes agglomérées aux particules.
La tension de surface
La tension superficielle, ou énergie d’interface, ou énergie de surface, est la tension qui existe à la surface de séparation de deux milieux. Cet effet permet par exemple aux insectes de marcher sur l’eau, à la rosée de ne pas s’étaler sur les pétales de fleurs, et explique la capillarité. La tension superficielle explique aussi la formation des bulles de savon.
Notion de tension interfaciale Du point de vue mécanique, lorsque deux phases sont en présence, tout se passe comme si elles étaient séparées par une membrane sans épaisseur uniformément tendue. Cette membrane fictive, qui remplace mécaniquement l’interface (phase volumique de faible épaisseur) est appelée « surface de tension ».
La condition de tension uniforme stipule que est indépendant de la direction de, et uniforme sur toute la surface. De même le travail nécessaire pour augmenter la surface de est donné par : Il existe différentes méthodes de mesure de la tension interfaciale. La plus simple est de mesurer la force nécessaire à l’arrachement d’un anneau d’une phase à l’autre dans le cas de 2 phases (liquide/liquide ou liquide/vapeur). Dans la littérature, on trouve une infinité de définition de la tension interfaciale. Nous proposons celle-ci : La tension interfaciale correspond au travail minimum « réversible » qu’il faut fournir pour amener des molécules (qui sont liées les unes aux autres par des forces de cohésion) du cœur du matériau ou d’une autre phase à sa surface afin d’augmenter la surface d’un incrément (ou accroissement), ou de la déformer. Cette énergie est rapportée à l’unité d’aire de surface ; elle s’exprime en joules par mètre carré (J/m2) (Andrieu et Müller 2005, Béranger et Mazille 2005).
La matière organique
La respiration est le principal moteur de la dégradation de la matière organique en milieu aquatique. La respiration est une réaction d’oxydo-réduction qui se traduit par l’oxydation du carbone organique en CO2. L’oxydation de la MO nécessite un accepteur d’électron. L’utilisation préférentielle de l’accepteur d’électron qui produit la plus importante quantité d’énergie libre lors de la dégradation de la MO est un modèle ancré en biogéochimie. Ceci explique la séquence verticale bien établie de réactions rédox sous l’interface eau sédiment ou dans une colonne d’eau stratifiée, ou encore la séquence temporelle des réactions rédox dans un milieu aquatique riche en carbone organique qui devient confiné. Ces séquences se traduisent par la réduction de l’oxygène en premier, suivie par la réduction des nitrates, des oxydes de manganèse, des oxydes de fer, des sulfates puis du dioxyde de carbone. Ces processus respiratoires microbiens prévus par la thermodynamique ont été démontrés. Ainsi, cette séquence thermodynamique permet de définir la réaction la plus favorable pour minéraliser la matière organique. In fine, la matière organique peut se conserver dans le milieu quand tous ces oxydants sont épuisés. Lorsque le carbone organique est oxydé en CO2, les autres composés de la matière organique sont minéralisés, en particulier l’azote et le phosphore organique qui sont transformés directement ou indirectement en ammonium et en phosphate dissous. A ces grands principes généraux gouvernés par la thermodynamique, de nombreuses études ont montré des nuances qu’il faut nécessairement prendre en compte pour comprendre les mécanismes qui mènent à l’eutrophisation. En effet, ces réactions dégagent de l’énergie. Cette énergie est mise à profit par des micro-organismes. Les communautés bactériennes et les différentes fonctions peuvent coexister. Ainsi, plusieurs processus peuvent se produire dans un même milieu, ou encore, l’ordre des réactions peut être nuancé par rapport à la séquence théorique de la thermodynamique. Citons quelques exemples : Il a été montré que la méthanogenèse se faisait en milieu côtier dès que le milieu était anoxique, même en présence de sulfate ou d’oxydes de fer ou de manganèse. L’utilisation des oxydes de fer comme accepteurs d’électron dépend grandement du pH et de la forme minéralogique de la phase FeIII . En effet, des processus de sulfatoréduction ou de méthanogenèse peuvent être favorisés dans des environnements riches en oxydes de fer. La taille des oxydes de fer joue aussi un rôle, les formes colloïdales étant plus oxydantes que les particules plus grandes. L’oxydation du carbone organique par les nitrates est plus favorable si l’azote est réduit en N2 que s’il est réduit en ammonium. Seule la première réaction devrait se produire. Pourtant la seconde a été mise en évidence dans tous les types d’environnements aquatiques. La nature même de la matière organique est un paramètre fondamental. Les formes les plus réfractaires ne sont pas minéralisées. Certaines formes ne le sont que par les oxydants les plus forts, l’oxygène en particulier. D’autres formes sont très labiles, en particulier la matière organique fraîche issue de la production primaire aquatique. Les formes labiles peuvent aussi jouer le rôle d’amorce pour favoriser la minéralisation de la matière organique réfractaire. Ce point important explique pourquoi la matière organique terrestre réfractaire ne s’accumule pas dans les proportions attendues à l’embouchure des fleuves. Cet effet d’amorce peut aussi permettre la minéralisation de matière organique préservée dans un milieu qui devient eutrophisé et où arrive de la matière organique labile, ce qui peut apporter un stock supplémentaire d’azote et de phosphore. La matière organique qui est minéralisée est complexe. L’état d’oxydo-réduction du carbone organique varie, ainsi que les proportions en éléments nutritifs majeurs tels que N, P, S, Si. Une vision plus complète des nutriments mis en jeu peut être donnée en remplaçant CH2O par (CH2O)x(NH3)y(H3PO4)z, où x, y et z représentent les proportions relatives de C, N et P dans la matière organique. Ces proportions sont en moyenne de 106:16:1 (rapport de Redfield) pour la matière organique phytoplanctonique. Elles sont très différentes pour la matière organique terrestre, par exemple 1158:24:1 pour des tissus racinaires, 1212:28:1 en moyenne pour les feuillages et 3007:45:1 pour les litières. Pour les plantes vasculaires aquatiques, le rapport C :N 😛 moyen est proche de 570 :29 :1. Lorsque la matière organique est minéralisée, le phosphore organique est minéralisé et libéré sous la forme d’ion phosphate. L’azote organique est libéré sous la forme d’ammonium. En milieu aérobie, l’ammonium est rapidement oxydé en nitrate. Tous les composés rédox décrits dans ces réactions peuvent interagir entre eux, générant des réactions secondaires. Ainsi, l’oxygène est un oxydant pour les composés réduits, tels que l’ammonium (processus de nitrification), Mn2+ et Fe2+ (précipitation d’oxydes de manganèse et de fer), les formes de soufre réduit (oxydation de H2S et HS en sulfate) ou encore le méthane (production de CO2). D’autres réactions rédox ont été décrites dans des travaux publiés. Par exemple, en présence d’oxygène, l’ammonium peut être oxydé directement en N2 par les oxydes de Mn sans que l’étape de nitrification ne se produise. Dans la plupart des environnements anaérobies, l’oxydant primaire du Fe2+ est le nitrate. Le nitrate peut également être réduit par le méthane ou les sulfures de fer. Le méthane dissous dans les eaux porales des sédiments est en grande partie oxydé par le sulfate. Il a été mis en évidence qu’il pouvait être oxydé par les oxydes de fer et de manganèse. Les oxydes de fer présentent aussi une grande variété. Il a été montré que seules les formes de Fe(III) les moins bien cristallisées étaient significativement réactives lors de l’utilisation de Fe(III) en tant qu’accepteur d’électron de la dégradation de la matière organique. Ces oxydes sont aussi les plus réactifs par rapport aux autres réducteurs, comme les sulfures. Les sulfures de fer peuvent être oxydés par les oxydes de manganèse. Enfin, les changements de pH et d’alcalinité dus à la production de CO2 et la réduction des sulfates génèrent la dissolution/précipitation des carbonates.
La pollution organique provient des effluents urbains insuffisamment épurés, mais également des rejets industriels, notamment du secteur agro-alimentaire. En se décomposant, les matières organiques et oxydables consomment une partie de l’oxygène nécessaire à la vie aquatique et provoquent l’eutrophisation (sans oxygène dissout) des eaux. Les dépôts de matières organiques mortes tendent par ailleurs à colmater le fond des rivières. Ces phénomènes contribuent à modifier les conditions de vie dans le milieu aquatique, ce qui entraîne la disparition des espèces les plus sensibles. Dans les estuaires la salinité varie en fonction des marées, les métaux complexés par les stations ERU sont relarguer au-delà de 5% de salinité, donc à chaque marée le milieu est exposé à des métaux à l’état libre.
Les éléments traces métalliques
Ce sont des micronutriments essentiels pour les organismes aquatiques, et se retrouvent naturellement dans l’eau à de faibles concentrations. Ils proviennent du lessivage du sol et du substrat rocheux. Les ions métalliques se trouvent en milieu aqueux sous diverses formes: libres, complexés avec des ligands simples organiques ou minéraux inorganiques, complexés avec des ligands macromoléculaires ou colloïdaux, adsorbés ou incorporés aux particules en suspension organiques ou inorganiques et adsorbés ou assimilés par les organismes. Les formes chimiques prises par les métaux sont importantes non seulement pour comprendre leurs toxicité et biodisponibilité, mais aussi, pour conceptualiser et évaluer les types de traitement des eaux. Leur importance a été clairement démontrée dans les traitements basés sur la coagulation, floculation ou adsorption utilisées, par exemple, pour éliminer les phosphates, fluorures ou la matière organique. D’autre part, les procédés utilisés pour le traitement des pollutions métalliques présentent généralement des rendements médiocres.
On appelle en général métaux lourds les éléments métalliques naturels, métaux ou dans certains cas métalloïdes caractérisés par une masse volumique élevée, supérieure à 5 g/ cm3. On retrouve dans certaines publications anciennes l’appellation de « métal pesant ». Quarante et un métaux correspondent à cette définition générale auxquels il faut ajouter cinq métalloïdes comme l’arsenic (As). Ils peuvent poser des problèmes de toxicité lorsqu’ils sont présents en trop grande quantité dans l’eau. Plusieurs métaux, dont notamment les métaux lourds, peuvent s’avérer toxiques même à l’état de traces. Les métaux les plus préoccupants, à cause de leur toxicité à long terme par accumulation dans les organismes et la chaîne alimentaire, sont le cadmium (Cd), le chrome (Cr), le cuivre (Cu), le mercure (Hg), le nickel (Ni), le plomb (Pb), et le zinc (Zn). Leur présence dans l’eau à des quantités préoccupantes est souvent associée à des rejets industriels ou agricoles.
De nombreux métaux et métalloïdes font l’objet de cycles biologiques. Ces biotransformations peuvent conduire à l’apparition de composés plus ou moins toxiques qui s’accumulent dans l’environnement.
Le terme spéciation est synonyme de distribution des espèces chimiques d’un élément, c’est à dire, sa distribution entre les différentes espèces chimiques y compris celles contenant des complexes dissous et des complexes avec particules. Cette définition est devenue confuse parce que ce terme est utilisé pour désigner non seulement la distribution entre ces diverses formes chimiques, mais aussi, l’ensemble des opérations analytiques qui permettent leur détermination (de la même manière que « extraction » dénote un procédé physique et une opération de séparation analytique): dans la suite de ce texte le terme spéciation est utilisé dans le sens analytique. L’approche analytique nécessite des méthodes pour déterminer la concentration des métaux totaux et des méthodes sélectives qui permettent de mesurer la concentration et certaines propriétés chimiques ou physiques des espèces. La sélectivité de ces méthodes est fréquemment dépendante de facteurs cinétiques, et on a besoin de définir les complexes cinétiquement inertes et labiles. Les complexes inertes sont ceux qui peuvent être séparés de leur milieu sans produire aucune modification autre que quantitative : si ces complexes participent à d’autres réactions dans le milieu étudié, la demi-vie de ces réactions doit être longue par rapport au temps de séparation et détermination analytique. C’est par exemple le cas des métaux adsorbés sur les solides ou incorporés dans les organismes: ces formes peuvent être séparés du milieu aquatique sans modifier sensiblement la quantité de métaux adsorbés ou assimilés. Dans certains cas, il existe des complexes inertes qui peuvent être isolés d’autres espèces du métal: par exemple les dérivés alkyls du Pb, Sn, As peuvent être séparés par chromatographique en phase gazeuse. Pour la plupart des complexes la sélectivité des méthodes de séparation n’est pas suffisante pour permettre l’isolement d’une espèce et, dans ce cas, on effectue un fractionnement en groupes de complexes homologues, complexes qui ne sont pas tous identiques mais qui possèdent des propriétés similaires. Les critères de fractionnement utilisés sont définis d’une façon opérationnelle, par exemple par la facilité avec laquelle les complexes sont : – décomposés soit par les acides ou réactifs oxydants, soit par réduction électrochimique, – retenus par une membrane filtrante, – ou bien adsorbés sur résines complexantes. Les complexes labiles devraient être idéalement séparés sans modifier le milieu original ou sans perturber les équilibres auxquels ils participent. Pratiquement, il existe très peu de méthodes suffisamment sensibles et sélectives pour atteindre cet objectif. De plus, la plupart de ces méthodes sont appliquées à la détermination des métaux libres et seulement à un nombre limité de métaux: électrodes spécifiques ioniques et polarographie impulsionelle directe ou à redissolution anodique. L’objectif primordial de la détermination des espèces métalliques vis-à-vis de l’environnement est d’identifier les espèces qui sont à l’origine des effets nocifs pour la vie aquatique, à partir de mesures de biodisponibilité et de toxicité. La toxicité se produit quand un organisme est incapable de faire face à une augmentation de la concentration de métal en l’utilisant directement, en le stockant ou en le sécrétant. La toxicité est fréquemment définie par les effets aiguës et les concentrations maximales admissibles ne tiennent pas compte des effets chroniques. L’interaction des métaux avec les compartiments intracellulaires est fortement dépendante des formes chimiques des métaux. Certaines espèces peuvent être capables de se lier avec des protéines extracellulaires et avec d’autres molécules biologiques ou bien de s’adsorber sur la paroi cellulaire ou encore se diffuser à travers la membrane cellulaire. A l’intérieur des cellules, les métaux peuvent s’assimiler ou perturber les réactions enzymatiques ou exercer d’autres effets toxiques. Ces effets sont très importants pour les petits organismes comme les algues et le plancton. La connaissance de la concentration totale d’un métal est souvent insuffisante pour évaluer tant sa toxicité que ses possibilités de transfert dans le milieu aquatique. Elle ne permet pas non plus de prévoir les effets des différentes filières de traitement sur ce métal. Les études toxicologiques ont montré que l’action d’un toxique sur le milieu dépend : de l’âge, de la taille et de la surface spécifique des organismes, de leur état physiologique, de la structure de la population, de la présence d’autres métaux ou de cations majeurs, de la température, du temps d’exposition, et principalement des formes physico-chimiques sous lesquelles ils s’y trouve. Dans les eaux de surface, les métaux lourds, au niveau des traces, sont présents sans différentes formes physico-chimiques. Ils peuvent être dissous ou particulatés (associés aux solides).
Prendre en considération, pour les métaux lourds dans le milieu aquatique les formes physico-chimiques suivantes : métaux particulaires, ce sont les formes retenues par un filtre de 0,45 mm (ex.: métaux liés aux sédiments), ions libres hydratés (ex.: Cd(H20)52 + ), complexes inorganiques simples (ex : Pb(H20)4), complexes organiques simples (ex.: Cu2+-glycinate), complexes inorganiques stables (ex.: PbS,ZnC), complexes organiques stables (ex.: Cu2+-fulvate), absorbés sur colloïdes inorganiques (ex.: Cu2+-Fe203), adsorbés sur colloïdes organique-inorganique (ex.: Cu2+-acide humique/Fe203). La variation des formes chimiques sous lesquelles se trouvent les métaux à l’état de traces peut changer leur biodisponibilité ou toxicité; par exemple la cobalamine (vitamine B12) est la seule forme assimilable du cobalt et les complexes chromeaminoacides sont les espèces assimilables du chrome. L’assimilation du fer par les algues vertes est précédée de la formation des complexes du Fe avec les exsudats des algues nommés siderochromes. Trois formes de transport des métaux à travers la membrane cellulaire sont reconnues : la diffusion passive, le transport impliquant des interactions avec des composants de la membrane et le transport actif dérivé du gradient de potentiel à travers la membrane. Certaines espèces peuvent se diffuser passivement à travers la membrane. Le degré de diffusion est fonction de la taille moléculaire des espèces métalliques, ainsi, les colloïdes sont virtuellement exclus, par contre, les ions libres se diffusent rapidement. Les ions métalliques libres hydratés semblent être les formes chimiques les plus toxiques (ex.: hydroxycomplexes de cuivre). La toxicité des métaux dépend en effet de la réactivité de chaque espèce chimique avec la membrane cellulaire des organismes et les ions bivalents sont les formes les plus favorisées (ex.: Cu(H20)42+, CuC1+, Cu(OH)+) . La pénétration des métaux dans la membrane cellulaire dépend de la solubilité des espèces métalliques dans les lipides: pour cette raison les espèces organométalliques neutres, par exemple, les dérivés alkyl du plomb, du mercure et de l’étain sont spécialement dangereux. La variation de l’état d’oxydation des métaux modifie leurs biodisponibilité ou toxicité: par exemple, Cr(III) est un élément essentiel mais le Cr(VI) est très toxique, As(III) est beaucoup plus toxique que As(V). Les ligands (donneurs d’électrons) organiques des eaux naturelles peuvent diminuer la toxicité des métaux par formation de complexes. Ces ligands comprennent les acides humiques, fulviques et tanniques. Ces acides sont polymères avec des fonctions acides, phénoliques et aminés. Un pH neutre favorise la formation de ces complexes et une diminution de la toxicité, c’est par exemple le cas du Pb et du Cu. Les conditions physico-chimiques du milieu peuvent modifier la toxicité des métaux lourds, par exemple, une augmentation de la salinité des eaux produit une diminution de la toxicité du cadmium dû à la formation des chlorocomplexes et à la diminution de la concentration du cadmium libre. Les complexes métalliques stables sont généralement considérés comme non toxiques. En outre ceci permet aux traiteurs d’eau de mieux définir les moyens à mettre en œuvre pour éliminer cette pollution métallique. (Flores-Rodriguez Julio Les Métaux Toxiques dans les Eaux Pluviales en Milieu Urbain)
La notion de coefficient de distribution, Kd. Ce coefficient est mesurable. Il est calculé par le rapport entre la concentration en métal particulaire par masse de suspensions et la concentration en métal dissous. Le terme particule, utilisé couramment dans la littérature, fait référence à des entités solides, vivantes ou non, dont la taille est supérieure à 0.45 um (voire 0.4 um). Bien que le choix de cette limite de taille découle d’une limitation expérimentale, elle se justifie essentiellement par le fait que la sédimentation affecte principalement les plus grosses particules et leur évolution dans l’estuaire. Les coefficients de distribution de métaux entre l’eau et les suspensions sont des paramètres essentiels et indispensables pour l’estimation des flux de métaux entrant aux océans à travers les estuaires. Il est bien connu que les particules en suspension dans les estuaires sont soumises aux processus de tri sédimentologique, associés à la décroissance des vitesses d’advection et entraînant le piégeage des métaux dans la zone côtière. La question qui se pose est alors : est ce que ces métaux restent définitivement piégés dans les sédiments ou bien vont-ils se désorbé ? Si désorption il y a, dans combien de temps aura-t-elle lieu? Immédiatement, dans quelques jours ou durera t-il des années ? Ces questions montrent d’une part l’importance des cinétiques de sorption et de désorption de métaux lors du transport de ces derniers dans les estuaires, d’autre part, mettent l’accent sur les problèmes de réversibilité des processus de sorption. Les mécanismes qui contrôlent les cinétiques des réactions de sorption sont très peu connus. En général l’adsorption d’ions métalliques sur les surfaces de particules peut être décomposée en deux étapes une réaction initiale rapide (quelques minutes) suivie d’un processus lent. L’étape rapide est souvent supposée être une réaction d’adsorption contrôlée par diffusion et prend quelques minutes pour atteindre l’équilibre quand la concentration de l’adsorbat au voisinage immédiat de la particule est constante. L’étape lente a été attribuée à des processus divers comme la précipitation à la surface ou la diffusion dans le micropore, ou la formation d’agrégat par floculation ou réarrangement structurel des espèces de surface, ou encore la migration à l’intérieur des particules vivantes. L’étude de la réversibilité des processus de sorption est de grande importance dans les estuaires. Dans le cas de réactions de sorption irréversibles, une partie des métaux est fixée définitivement sur les particules et n’est plus échangée quelque soient les changements de la composition de l’eau.
Le fer rencontré en milieu aquatique n’est en général pas considéré comme un élément toxique. Cependant, il est suffisamment abondant et réactif pour influencer le devenir des éléments traces métalliques. Il est présent dans l’eau avec une concentration très faible qui peut augmenter suite à la lixiviation des terrains riches en fer ou à cause d’une pollution industrielle. Le fer se trouve dans l’eau sous une forme dissoute ou en solution colloïdale.
Il se présente un danger de toxicité assez modérée. Pour la vie aquatique, cette toxicité est difficile à préciser car elle est fonction de l’état chimique du métal et aussi de la présence du précipité d’hydroxyde de fer qui tend à se déposer sur les branchies de poissons, et entraîner leur colmatage. Pour l’eau de boisson, l’OMS (Organisation Mondial de la Santé) donne une DMA (Dose Maximale Admissible) de 1mg/L et une DMS (Dose Maximale Souhaitable) de 0.1mg/L. Il favorise aussi la prolifération bactérienne.
Les eaux de rejets de stations émettent des quantités de chlorure ferrique en excès pour abattre les matières en suspensions (3 à 4 mg/L de rejet dans l’effluant). Le problème est que le chlorure ferrique est toxique pour l’environnement et décime la biodiversité notamment bactérienne en aval des stations. Ce qui s’observe par une couche de bio film sur le lit des milieux récepteurs, qui provoque une asphyxie des boues ou ce trouve la majorité de la vie phytoremédiatrice.
Le sélénium
Communément appelé silice dissoute (Sid). Il s’agit d’un des nutriments les plus importants des écosystèmes continentaux. La silice dissoute est également l’un des nutriments majeurs pour les systèmes marins. Son origine n’est pas anthropique et sa présence est essentiellement liée aux flux provenant du bassin versant. Le cycle de la silice est plus simple que celui des autres nutriments. Sa présence sous forme dissoute est liée à l’altération des roches silicatées du bassin versant mais également à la solubilisation des tests silicatés d’organismes utilisant la silice dissoute pour leur croissance (i.e. les diatomé). La présence de silice dissoute est essentielle au développement de diatomées. La présence de diatomées est associée à une bonne qualité de l’eau et permet notamment de favoriser la biodiversité du milieu. D’autres végétaux aquatiques se servent également de la silice dissoute, notamment dans l’élaboration de leur paroi. Le rapport entre l’azote et la silice peut être déterminant dans le développement d’organismes sensibles à la présence de silice. Des rapports Sid/DIN supérieurs à 1 ont été corrélés avec une meilleure croissance des diatomées par rapport aux flagellés dans des eaux côtières.
Le nickel peut également être absorbé par voie orale. La pénétration cutanée est faible. Toxicocinétique : absorption, transport, distribution, élimination, après absorption par voie pulmonaire ou digestive, le nickel diffuse dans tout l’organisme puis il est rapidement excrété par voie urinaire, ce n’est pas un toxique cumulatif. Mécanisme d’action, le métal se fixe préférentiellement au niveau du rein et du foie. Ce métal est allergisant. Le nickel sous certaines formes, est également cancérogène (fosses nasales, poumons). Dans l’eau et en conditions naturelles, le zinc se trouve avec de très faibles concentrations, inférieures à 5mg/l. Mais, ces concentrations peuvent augmenter suite à des lâchers de zinc, si des canalisations de l’étain ou de fer galvanisé sont attaquées par les eaux riches en chlorure et en sulfate. Le zinc présente une toxicité relative pour les organismes aquatiques à partir de quelques mg/l et ceci en fonction de la minéralisation de l’eau et de l’espèce considérée. Pour l’homme, la quantité tolérable est limitée par un goût désagréable obtenu à des concentrations de l’ordre de 5-15mg/l. Et puisque le zinc est souvent couplé au cadmium au chlore, la dose est abaissée à 1mg/l.
Notamment Le chlorhydrate d’aluminium appartient à une famille de composés chimiques que l’on trouve dans les antisudorifiques (sulfate d’aluminium, aluminate de sodium ou des polymères aluminiques) et les agents de floculation utilisés dans le traitement de l’eau. Ces agents de floculation sont déverser dans les bassins de stations ERU avec des pompes péril statique, mais les variations de concentration dépendent du flux d’entré. Donc le problème est qu’un surdosage provoque une pollution de l’environnement, en outre les normes de rejets l’analyses ne mesure pas l’aluminium. Les effluents contiennent 3 à 4 mg/L d’Al. À l’instar d’autres sources d’exposition au métal, il fait régulièrement la manchette en raison d’un lien hypothétique entre l’aluminium et des maladies d’origine neurotoxique, comme la maladie d’Alzheimer. L’accumulation de l’aluminium est un problème majeur chez l’insuffisant rénal. L’aluminium entre en compétition au niveau cellulaire avec le calcium. La forte fixation cérébrale de l’aluminium est responsable de la symptomatologie clinique (neurotoxicité). Parmi les mécanismes évoqués, on note : la modification de la barrière hématoencéphalique ; la production d’oxygène radicalaire ; la fixation sur les acides nucléiques. (Jean pierre GOULLE praticien pharmacotoxicologie Havre)
Lemercure: il subit une méthylation essentiellement par les bactéries sulfato-réductrices anaérobies, par transformation en méthylmercure (MeHg), très facilement bioaccumulable. Cette biotransformation est favorisée par des valeurs faibles du pH, ainsi que par des concentrations élevées en matière organique.
Le mercure présente un risque majeur pour l’écosystème marin et pour le consommateur humain, en raison de sa toxicité et de ses capacités de biomagnification par la chaîne alimentaire. Il est utilisé dans des activités très variées (industrie papetière, industrie de bois, industrie chimique, agriculture…). Dans l’environnement, les composés méthylés ont une place particulièrement importante dans les cycles biogéochimiques. La méthylation du mercure a été constatée dans les sédiments sous l’action des micro-organismes et dans la colonne d’eau en présence de phytoplancton.
Hg minéral® Méthylmercure (Hg CH3) ® Diméthylmercure (GLS mémotec19)
Le mercure ainsi formé à une très grande faculté de biomagnification dans les chaînes alimentaires, et sa proportion augmente progressivement quand on passe d’un échelon trophique au suivant. Les effets toxiques de méthylmercure se manifestent par des lésions du système nerveux central, spécialement les centres sensoriels de la vision, de l’audition et de la coordination altérés. Une exposition plus longue entraîne une ataxie (incoordination motrice) et des perturbations de la vision, débilité, paralysie puis décès.
Le plombonstitue 0,014 % de la masse de la croûte terrestre, ce qui le place parmi les métaux industriels, entre le cuivre (0,035 %) et l’étain (0,002 %). Il se trouve dans l’eau de mer principalement sous forme de carbonates PbCO3 (40 à 80 %) ou de chlorures PbCl2 (1 à 40 %) et PbCl+ (2 à 19 %).
La pollution par le plomb a plusieurs origines parmi lesquelles les industries (fonderies de la métallurgie, les câbles, bâtiments..), les incinérations d’ordures et les peintures réalisées avec des composés de sulfates et de plomb basique. En plus, le plomb est utilisé comme agent de traitement de certaines maladies en agriculture. Généralement la contamination des sédiments décroît de la surface vers les horizons profonds, traduisant ainsi les effets de l’ère industrielle. Les niveaux de présence sont de l’ordre de 5 à 20 µg/g dans les dépôts industriels, 50 à 100 µg/g sur le plateau continental et peuvent dépasser 150 µg/g en milieu côtier. Plus la chaîne du plomb est importante, plus il est toxique. Le plomb sous forme de tetraéthyl-plomb est infiniment plus élevé que celle du plomb, et le tetraalkyl de plomb est le plus toxique.
Pour les organismes aquatiques, le plomb présente une toxicité létale lors des concentrations comprises entre 2,46 et 8,8 mg/l. Sa concentration inhibant le développement embryonnaire de la moule dépend de la salinité et de la température. Les concentrations inhibitrices les plus fortes sont de l’ordre de 500 mg/l et elles sont notées aux conditions optimales de 15,6°C avec une salinité de 35 p.1000. Pour le plomb inorganique, les concentrations à partir desquelles des effets sublétaux peuvent être observés sont comprises entre 0,5 µg/l pour la croissance du phytoplancton et 500 µg/l pour l’apparition d’anomalies dans le développement embryonnaire des bivalves.
A l’état naturel du cadmium est presque toujours associé à d’autres métaux (zinc et plomb). Sa teneur dans l’eau va de traces non détectables à 130 mg/L. Le cadmium est principalement utilisé pour la fabrication des batteries, mais son introduction dans le milieu marin peut résulter de l’activité minière. Il est également utilisé comme matériel de contrôle ou de protection dans les centrales nucléaires, car il absorbe facilement les neutrons de faible énergie. La présence du cadmium dans le milieu aquatique a un impact sur ses organismes, mais sa toxicité diffère selon l’espèce et la concentration. La concentration en cadmium dans la chaîne alimentaire double tous les 20 ans (en raison du transfert facile du cadmium du sol vers les végétaux et de la très longue demi-vie de cet élément dans les milieux biologiques). La concentration en cadmium dans le rein de l’homme est 50 fois plus élevée qu’au début du 20e siècle. Le cadmium induit chez l’homme la synthèse hépatocytaire d’une protéine : la métalothionéines (MT), puis la formation d’un complexe Cd-MT, enfin l’accumulation du complexe Cd-MT dans le cortex rénal. La MT neutralise les effets toxiques du Cd, mais, si la teneur du complexe Cd-MT est supérieure à 180 µg/g de cortex, il forme un complexe Cd-MT avec d’autres ligands qui entraîne une atteinte rénale irréversible : d’abord une protéinurie tubulaire modérée peut être constatée, puis une atteinte rénale plus sévère, avec hypercalciurie, glycosurie, phosphaturie, amino-acidurie, peut s’installer. Le cadmium présentant une affinité pour les groupements thiols est à l’origine de nombreuses perturbations métaboliques et enzymatiques. Les mécanismes d’action pourraient également impliquer une interférence entre le métabolisme de certains métaux, et même du calcium. La dynamique du mercure dans l’environnement est conditionnée par ses propriétés physiques (volatilité à température ambiante), ses propriétés chimiques (stabilité de ses liaisons avec le carbone et le soufre) et enfin ses propriétés biologiques (très forte bioconcentration et sa toxicité). Le mercure ne fait pas partie des oligo-éléments, il n’a aucun bénéfice biologique avéré. Le mercure est toxique dans la plupart de ses formes chimiques. La forme méthylée est la plus toxique. Chez les vertébrés, le méthylmercure est un neurotoxique puissant qui entraîne des lésions au niveau du cerveau et du système nerveux central. Il provoque des tremblements, des pertes d’équilibre, des paralysies, l’inhibition du développement cérébral du fœtus, etc. Le méthylmercure passe facilement les barrières biologiques, lui donnant ainsi la capacité de se bioaccumule et de se bioamplifie le long de la chaîne trophique. Même si le méthylmercure est présent à de très faibles concentrations dans l’eau, il peut se concentrer jusqu’à 10 millions de fois dans les organismes aquatiques comme les carnassiers. L’exposition des humains au méthylmercure est principalement contrôlée par la consommation des poissons. Les poissons de haut niveau trophique peuvent présenter de très fortes concentrations en méthylmercure. La teneur maximale tolérable pour la vente est de 1mg.kg-1 poids frais suivant la commission européenne du 19 mai 1993 93/351/EEC. L’organisation mondiale de la santé (OMS) recommande aux femmes enceintes de limiter leur consommation de poissons sous risque de retards psychomoteurs de l’enfant. Le mercure est un élément chimique de symbole Hg, appartenant à la famille des métaux lourds. Sa masse atomique est de 200,59 g/mol. C’est le seul métal à l’état liquide dans les conditions normales de température et de pression de surface. Il existe sept isotopes stables naturels du mercure, de masses 196, 198, 199, 200, 201, 202 et 204. Il est présent dans tous les compartiments terrestres (hydrosphère, atmosphère, lithosphère et biosphère) sous différentes formes chimiques. Sous sa forme élémentaire le mercure est très volatile. Cette propriété lui permet d’avoir un temps de résidence atmosphérique de l’ordre d’une année. Ainsi, sa forme gazeuse peut être redistribuée à l’échelle planétaire par la voie atmosphérique. Il possède deux autres degrés d’oxydation. Les composés mercureux (Hg(I), forme inorganique), ont une faible stabilité et sont donc peu présents dans l’environnement. Les composés mercuriques Hg(II), forme inorganique forment des composés stables avec le carbone, l’azote, le chlore, le brome, l’iode et surtout avec le soufre et le sélénium. Dans les milieux aquatiques, Hg peut former des chlorocomplexes, des thiocomplexes ou des complexes organiques (humiques et fulviques). La prédominance de l’un ou l’autre de ces complexes dépend de la concentration des ligands présents dans chaque type d’environnement. Dans les eaux côtières, les complexes organiques et les thiocomplexes sont les espèces dominantes de mercure par rapport aux complexes inorganiques. L’affinité du mercure pour la matière organique dissoute et particulaire tient surtout du fait de la présence de soufre. Le mercure se démarque des autres métaux par une forte tendance à former des liaisons covalentes plutôt que ioniques, notamment avec le carbone, le soufre et le sélénium. Il en résulte la formation de composés méthylés du mercure (organo-mercuriels), qui sont les formes les plus toxiques et bioaccumulables, et de composés de sulfure et de séléniure de mercure, plus stables et insolubles. La méthylation du mercure inorganique n’a lieu qu’en milieu aqueux. Cette méthylation peut être induite par le biota lorsqu’il y a absorption, passive ou active d’Hg(II) par certains microorganismes qui, en réponse, sont capables de le méthyler, c’est-à-dire de créer des liaisons covalentes entre Hg(II) et deux groupements méthyl. La forme d’Hg (II) la plus disponible pour ces microorganismes serait la forme HgS (aq). La forme monométhylée est celle qui s’accumule dans le milieu. Elle est issue de la décomposition abiotique du diméthylmercure (DMHg) en monométhylmercure (MMHg). Les organismes les plus aptes à méthyler le mercure sont principalement les bactéries sulfato-réductrices (BSR) mais aussi, dans une moindre mesure, les bactéries ferri-réductrices et méthanogènes. Cette méthylation microbienne est localisée dans la zone anoxique des sédiments ou de la colonne d’eau. La méthylation abiotique est également possible, mais peu observée. Le MMHg peut être déméthylé en Hg(II) ou Hg0. Cette décomposition peut être photochimique (abiotique) ou biotique, par l’intermédiaire de certains microorganismes par voie réductrice ou oxydative. Plusieurs études ont aussi mis en évidence une activité de méthylation du mercure au niveau du périphyton associé aux racines et aux feuilles de macrophytes aquatiques. Les plantes aquatiques fournissent un support physique riche en composés organiques pour le développement de biofilms où peuvent croître des microorganismes anaérobies. Là, le mercure peut s’adsorber et être méthylé, créant ainsi une source de MMHg pour la chaîne alimentaire.
Le comportement du cadmium dans les estuaires est bien documenté. Les travaux menés sur cet élément ont systématiquement montré un enrichissement marqué dans la phase dissoute. Cet enrichissement a généralement été attribué à la désorption du métal depuis les particules en suspension. Cette désorption est liée à l’augmentation de la force ionique au cours du transit estuarien des particules. Les expériences de Paalman (1994) indiquent que cette désorption est le résultat d’une compétition entre les ions Ca2+ et les ions Cd2+ sur les sites d’adsorption des oxydes de fer et de manganèse. Les calculs de spéciation ont suggéré que cette désorption est favorisée par la formation de chlorocomplexes. D’autres travaux ont par contre montré l’implication des ligands organiques dans la complexation du cadmium dissous. Lors du transit estuarien, il existe une désorption importante du métal depuis les particules en suspension. L’enrichissement en métal dissous qui en résulte se fait essentiellement à la faveur de composés de faible poids moléculaire Nos travaux ont également permis de mieux cerner les facteurs qui contrôlent cette désorption du métal ; un temps de résidence des eaux réduit ainsi qu’un bouchon vaseux concentré favorisant une désorption à des salinités plus élevées.
Par ingestion alimentaire d’arsenic : c’est le mode le plus fréquent d’exposition dans la population générale. Après le plomb et le mercure, l’arsenic est le troisième poison majeur de l’environnement. La contamination par l’arsenic minéral des nappes phréatiques et de l’eau constitue un problème grave de santé publique dans certaines régions du globe où sévit un arsénicisme endémique. L’absorption digestive est pratiquement totale pour les différentes formes d’As (As3+, As5+, As organique). Après transport sanguin par les protéines, l’arsenic se fixe fortement sur les globules rouges, le foie, les reins, la rate et les phanères (cheveux, poils, ongles). Alors que l’As organique est éliminé dans les urines en l’état, l’As minéral est éliminé sous forme de dérivés méthylés (80 %) et de dérivés inorganiques (20 %). Les dérivés minéraux de l’arsenic sont doués de propriétés cancérogènes et mutagènes. Les dérivés organiques de l’arsenic (arsénobétaïne en particulier), contenus en quantité non négligeable dans certains aliments (chair de poisson, crustacés), sont beaucoup moins toxiques que les dérivés minéraux de l’arsenic. Des travaux menés se sont intéressés à la spéciation inorganique de l’arsenic dans l’estuaire de la Penzé. Dans cette étude, les teneurs relativement élevées en As inorganique dans les eaux fluviales (supérieures à 10 nM) ont été reliées à la nature fortement agricole du bassin versant, en particulier à l’utilisation de ce métalloïde comme agent antibiotique pour l’élevage porcin. Dans la zone du gradient salé, l’arsenate [As(V)], espèce majoritaire, présente un comportement non conservatif (Figure I-10). Le retrait de cette espèce de la fraction dissoute se fait par coprécipitation avec les oxydes de fer. L’arsenite [As(III)] présente quant à lui un comportement conservatif. Le fait que l’arsenate soit la seule espèce affectée par la coprécipitation implique que la part d’arsenite augmente (de 5 à 15%) lors du transit estuarien.
Concernent l’agglomération de Lannion, des rejets d’arsenic sont présent dans l’eau : Dans les années 50 des forages de prospection d’uranium on été réaliser, ce qui à perforer la gangue de quartz et libérer de l’arsenic dans la rivière à 800 m en aval du captage d’eau potable.
L’exposition à l’arsenic est un problème de santé mondial. Ce métalloïde toxique est omniprésent dans l’environnement et contamine les aliments et l’eau potable. Une fois ingéré, il subit un processus métabolique complexe dans le corps, ce qui contribue à son accumulation et à sa réactivité. La toxicité de l’arsenic découle de l’induction d’un stress oxydatif, de l’inhibition des protéines contenant des thiols et du mimétisme des phosphates inorganiques. L’empoisonnement à l’arsenic est associé au développement de troubles de la reproduction. Chez les mâles, l’arsenic provoque une réduction du poids des testicules et des altérations de la stéroïdogenèse et de la spermatogenèse. De plus, il réduit le nombre et la qualité des spermatozoïdes récoltés dans la queue de l’épididyme. Les mitochondries sont des cibles de la toxicité de l’arsenic en raison de la production de radicaux libres et de leur forte teneur en protéines riches en cystéine et en acides gras. Le dysfonctionnement mitochondrial peut contribuer aux troubles de la reproduction car cet organite est crucial pour contrôler les événements testiculaires et épididymaires liés à la production et à la maturation des spermatozoïdes. Toutes ces altérations médiées par l’exposition à l’arsenic contribuent à l’échec de la compétence reproductive masculine en réduisant la viabilité des gamètes. Cette revue décrit les mécanismes potentiels de la toxicité de l’arsenic, ses effets néfastes sur les organes reproducteurs mâles et ses conséquences sur la fertilité des spermatozoïdes. Le dysfonctionnement mitochondrial peut contribuer aux troubles de la reproduction car cet organite est crucial pour contrôler les événements testiculaires et épididymaires liés à la production et à la maturation des spermatozoïdes. Toutes ces altérations médiées par l’exposition à l’arsenic contribuent à l’échec de la compétence reproductive masculine en réduisant la viabilité des gamètes. Cette revue décrit les mécanismes potentiels de la toxicité de l’arsenic, ses effets néfastes sur les organes reproducteurs mâles et ses conséquences sur la fertilité des spermatozoïdes. Le dysfonctionnement mitochondrial peut contribuer aux troubles de la reproduction car cet organite est crucial pour contrôler les événements testiculaires et épididymaires liés à la production et à la maturation des spermatozoïdes. Toutes ces altérations médiées par l’exposition à l’arsenic contribuent à l’échec de la compétence reproductive masculine en réduisant la viabilité des gamètes. Cette revue décrit les mécanismes potentiels de la toxicité de l’arsenic, ses effets néfastes sur les organes reproducteurs mâles et ses conséquences sur la fertilité des spermatozoïdes.
Plus de 60% de la production de chrome est réservée à la fabrication des produits métalliques, et environ 20% est utilisé dans les réfractaires. Il est principalement utilisé dans les alliages avec le fer, le nickel ou le cobalt. L’impact immédiat de sa présence dans le milieu marin se manifeste par sa concentration dans les différents organismes aquatiques. Les organismes, même loin des zones polluées, concentrent le chrome dans leurs cellules.
Tout substrat ou structure en immersion dans l’eau de mer représente un support pour la colonisation d’espèces sessiles mais aussi motiles. La fixation des salissures (ou fouling) sur les coques de bateau s’organise dans un ordre défini. Dans un premier temps, les molécules organiques (polysaccharides, protéines …) sont rapidement accumulées sur la surface. Dans la foulée, les bactéries et les diatomées unicellulaires prolifèrent et adhèrent sur ce nouveau substrat pour former un biofilm. L’irrégularité du biofilm et sa grande richesse en nutriment permet l’adsorption de plus grosses particules et d’organismes tels que des spores d’algues, des protozoaires ou des cyprides de barnacle. Cette colonisation d’organismes plus complexes est la troisième étape du développement du biofouling. La phase finale correspond à la croissance de macroorganismes et à la fixation d’invertébrés marins ainsi que des algues. L’utilisation de protection efficace contre le développement de biosalissures à la surface des coques de bateau a toujours été une préoccupation pour les navigateurs. Les premiers navigateurs recouvraient leurs coques en bois de produits naturels tels que la cire ou le goudron. Selon les mêmes sources, les Phéniciens et les Carthaginois semblent avoir été les premiers à utiliser le cuivre et le plomb afin de protéger leurs bateaux. Entre le 13ème et le 15ème siècle, le goudron fut très largement utilisé pour protéger les coques et mélangé parfois à de l’huile, de la résine ou du suif. La première utilisation de revêtement de cuivre authentifiée a été signalée sur le bateau de guerre anglais l’HMS Alarme en 1758. Son succès relatif a encouragé l’utilisation du cuivre sur d’autres bateaux (WHOI 1952). A partir de 1960, une technique de copolymère autopolissante révolutionnaire à base de tributylétain (TBT) a été utilisée. Le TBT devint très vite la molécule la plus utilisée car très efficace et peu coûteuse. Cette utilisation représentait dans les années 1980 près de 70% de la consommation mondiale d’étain. Au début des années 1980, plusieurs pays comme la France et le Royaume Uni ont commencé à réguler l’utilisation des TBT dans les peintures antifouling. Suite aux découvertes des dégâts causés par le TBT sur les huitres notamment d’autres pays ont commencé à réguler les peintures. En 1998, Alzieu a estimé qu’une coque de bateau relarguait entre 1 et 10 µg de TBT/cm²/jour afin d’assurer une protection contre le biosalissures. Ceci correspond à 0.2-2 g/jour pour un petit bateau et jusqu’à 50-500 g/jour pour les bateaux de marchandises. En 1990, l’Organisation Maritime Internationale (OMI) des Nations Unis a adopté une résolution recommandant aux gouvernements d’adopter des mesures restrictives envers l’utilisation des TBT dans les peintures. Aussi, l’usage du TBT en tant que biocide fut interdit en 2008. Il est à noter qu’en 2008 le Sénat américain a donné son autorisation pour la ratification de la convention toutefois, en août 2009, le Congrès n’avait toujours pas permis cette ratification. Ainsi, l’utilisation de peintures antifouling au TBT aux Etats Unis n’est toujours pas considérée comme un délit. En 2003, l’Union Européenne a interdit l’application extérieure de peintures au TBT pour tous les bateaux battant pavillon de l’Union à l’exception des bateaux militaires. Depuis 2008, les systèmes antisalissures susceptibles de libérer des organostanniques sont totalement proscrits, et il est obligatoire d’éliminer les revêtements des coques de bateaux contenant du TBT sauf s’ils sont recouverts d’un revêtement « barrière ». Malgré ces interdictions, le TBT, est responsable aujourd’hui encore d’une contamination persistante des sédiments en raison de leur faible degré de dégradation, suivant les conditions, qui peut s’avérer toxique pour les écosystèmes aquatiques. Aujourd’hui, le cuivre sous sa forme CuO ou CuCNS (thiocyanate de cuivre) est redevenu la base des antifouling mais d’autres biocides sont venus compléter son action. C’est ainsi que le zinc pyrithione (C10H8N2O2S2Zn), le cuivre pyrithione (C10H8N2O2S2Cu), le SeaNine 211 (C11H17Cl2NOS) ou l’Irgarol 1051 (C11H19N5S) sont largement utilisés dans les formulations des nouveaux antifouling. Ces produits chimiques organiques sont utilisés alors que leurs comportements, leurs toxicités et leurs effets sur l’environnement aquatique sont relativement mal connus. Les effets sur les organismes du Cu et du TBT Il a été montré que la contamination métallique due aux peintures antifouling réduisait la biodiversité des écosystèmes marins et pouvait favoriser le développement d’espèces non indigènes plus tolérantes aux métaux que les espèces indigènes. C’est un point crucial dans les ports où la contamination est importante et où les espèces non indigènes peuvent être importées, attachées aux coques de bateaux. Une étude de Piola & Johnston (2007) a montré que les espèces non indigènes transportées sur les coques et qui avaient subi une forte pression de sélection liée à la présence de peinture antifouling à base de Cu étaient plus tolérantes aux métaux que les espèces indigènes. Des effets directs peuvent être observés sur les organismes aquatiques exposés aux contaminants via les eaux, les sédiments et la nourriture. Le Cu est un élément essentiel pour les organismes mais peut s’avérer toxique lorsque ses concentrations dépassent les simples besoins nutritionnels. Pour de nombreuses espèces aquatiques, par exemple chez les mollusques, les crustacés, les poissons et les mammifères l’accumulation du cuivre est régulée. Les entrées de cuivre se font préférentiellement sous forme ionique (Cu2+) par des protéines de transport membranaire. Les bivalves accumulent le cuivre principalement dans l’hépatopancreas, les gonades et les branchies. C’est au niveau des hépatocytes que le cuivre va provoquer le plus de dysfonctionnement voir des lyses chez les macroorganismes benthiques. Plusieurs études ont démontré la toxicité du Cu pour de nombreux organismes marins même à de faibles concentrations. Le Cu relargué à partir de particules de peinture antifouling à une concentration de 4 mg L-1 réduit l’activité photosynthétique de l’algue Ulva Lactuca. Une inhibition de la croissance des diatomées par le Cu a été observée à des concentrations extrêmement faibles. Dans les sédiments, la diversité faunistique est réduite lorsque les concentrations en Cu dépassent 30 µg g-1. Le TBT est un composé organométallique. Dans l’eau la molécule est présente sous forme cationique (TBT+) ou sous forme d’une particule neutre (TBT-OH ou TBT-Cl). La molécule de TBT se dégrade dans l’environnement via des réactions photochimiques (dégradation aux UV…) ou liée à l’activité microbiologiques (déalkylation…) en dibutylétain (DBT), puis monobutylétain (MBT) et enfin en étain inorganique. Le temps de demi-vie du TBT dans l’eau de mer est de six jours à quelques mois tandis qu’il varie de un an à plusieurs décennies dans les sédiments en lien avec les conditions du milieu. Le TBT est une molécule hydrophobe avec un coefficient octanol-eau (Kow) compris entre 5000 et 7000. Les organismes peuvent donc bio concentrer le TBT dans leurs tissus comme il a été démontré chez Daphnia magna ou Chironomus riparus. L’extrême toxicité des OSn pour les communautés biotiques a été démontrée pour de nombreuses espèces in vivo et in vitro. De nombreuses études écotoxicologiques montrent la contamination de plusieurs organismes quelles que soient leurs places dans le réseau trophique de l’écosystème aquatique. Les OSn deviennent toxiques pour certaines espèces à partir de concentrations comprises entre 1 et 10 ng Sn L-1. Les gastropodes marins et les huitres sont les organismes les plus sensibles aux effets des OSn, tandis que les poissons peuvent supporter des concentrations jusqu’à 100 ng/L. Les TBT et autres composés organostanniques agissent sur plusieurs fonctions différentes. Ainsi, ils perturbent l’homéostasie du calcium, inhibent la phosphorylation mitochondriale et la synthèse d’ATP ou inhibent la photophosphorylation des chloroplastes. Ils sont génotoxiques et endommagent l’ADN. Les OSn sont aussi des perturbateurs endocriniens. Ils inhibent les enzymes CYP et en particulier des aromatases responsables de la conversion de la testostérone en œstradiol causant la masculinisation des gastropodes femelles et de près de 100 autres espèces. Les femelles développent des organes reproducteurs mâles à partir de concentrations en TBT de l’ordre du ng/L. La problématique des OSn bien qu’identifiée dès les années 1980 est toujours d’actualité dans la plupart des environnements côtiers. Leur faible dégradation en fait un polluant persistant et le TBT a été caractérisé par Goldberg (1986) comme la molécule la plus toxique ayant été délibérément introduite dans le milieu aquatique.
Identification des sources d’éléments traces La directive cadre sur l’Eau (DCE, 2008/105/CE) concernant les eaux côtières (eaux de transition) impose aux États membres « d’améliorer les connaissances et les données disponibles sur les sources des substances prioritaires et les voies de pollution afin d’identifier des options de contrôles ciblés et efficaces ». La directive cadre stratégie pour le milieu marin (DCSMM, 2008/56/CE) impose l’obtention ou le bon état écologique des eaux côtières et marines à l’horizon 2020. Dans le cadre du Grenelle de la Mer, le Groupe de Travail n°11 « sédiments de dragage » a émis un certain nombre de recommandations, notamment sur la réduction des risques de contamination des sédiments marins, en agissant, en amont, sur les principales activités polluantes, à l’échelle des bassins versants. L’application des différentes directives et la prise en compte des recommandations formulées par le Grenelle de la Mer posent le problème de l’identification des sources de pollution. Pour cela, le développement d’outils de traçage des sources comme les isotopes stables est nécessaire.
(Selon Virginie Tanguy UBO Spéciation colloïdale des éléments traces métalliques en milieu estuarien) La quantification des flux de métaux pour les différentes fractions de taille de colloïdes a permis de mieux cerner les principaux échanges qui s’opèrent au sein de l’estuaire. Pour le plomb, il existe essentiellement un enrichissement de la fraction particulaire dans la zone de mélange ; celle-ci est liée à un transfert du métal depuis la fraction dissoute. La floculation des oxydes de fer, phénomène identifié comme étant responsable de ce transfert, agit principalement sur la fraction colloïdale de haut poids moléculaire. La perte de métal dans cette fraction est de l’ordre de 80% ; dans la fraction de plus faible poids moléculaire, elle est d’environ 50%. Pour ce métal, la fraction réellement dissoute n’apparait que très peu affectée par le transfert du métal dans le gradient salé estuarien. Dans le cas du cadmium, nos estimations montrent qu’il existe un transfert important du métal de la phase particulaire vers la phase dissoute. Ce transfert se fait essentiellement à la faveur des colloïdes de bas poids moléculaire et de la fraction réellement dissoute. La perte en cadmium dans la fraction particulaire est d’environ 30% lors du mélange estuarien. Dans le même temps, un facteur d’enrichissement d’environ 20 environ est constaté pour les deux fractions en solution. Pour le cuivre, nos résultats indiquent, d’une manière globale, que le métal est transféré de la phase particulaire vers la phase colloïdale lors du mélange des eaux dans l’estuaire. La « perte » en cuivre particulaire est d’environ 25%. Parallèlement à celle-ci, un enrichissement, avec des facteurs d’augmentation compris entre 1,5 et 2, est constaté dans les différentes fractions colloïdales ainsi que dans la fraction réellement dissoute. Ces travaux menés dans l’estuaire de la Penzé ont donc montré une présence particulièrement importante des éléments métalliques (Cu, Pb, Cd) dans la fraction colloïdale. Si le cadmium est trouvé dans la partie plutôt basse du spectre de taille colloïdal (300 kDa) ; le cuivre présentant quant à lui une distribution intermédiaire. Lors du cycle saisonnier, deux phénomènes majeurs ont été identifiés. Ils affectent non seulement la répartition colloïdale des éléments étudiés mais jouent également un rôle important dans les quantités et les formes transférées vers la zone côtière. Ces deux phénomènes sont : – D’une part, la forte mobilisation des substances humiques depuis les sols du bassin versant lors des évènements pluvieux intenses de l’automne. – D’autre part, la dégradation du matériel organique particulaire d’origine continentale dans une zone très localisée du compartiment benthique au printemps.
Sont une famille de composés est très réactive dans les milieux naturels, ils ce combinent très facilement sur tous types de polluants organiques qui forment les méthabolites, ce qui les rendent difficilement détectables par l’appareillage utiliser en laboratoire.
Les chlorures sont très répandus dans la nature sous forme de sels de sodium (NaCl), de potassium (KCI) et de calcium (CaCI2). La présence de l’ion chlorure sous les formes C1–, ClO–, ClO2–ou ClO3– dans l’eau, peut avoir diverses origines, telles que le passage à travers des terrains salés, les industries extractrices et dérivées (soudières, salines, mines de potasse, industries pétrolières…), la dissolution de dépôts de sel gemme, etc. Les eaux chlorurées alcalines sont laxatives. Généralement les chlorures présents dans l’eau destinée à la consommation humaine n’ont pas de conséquences toxiques pour l’homme. Cependant, ils peuvent être préjudiciables aux personnes atteintes de maladies rénales ou cardio-vasculaires. Ils peuvent favoriser la mobilisation d’ions indésirables ou toxiques à partir des canalisations métalliques, surtout si celles-ci véhiculent de l’eau chaude.
Le chlore Cl2 est un élément qui provient essentiellement de l’industrie chimique. Sa présence dans le milieu marin a un impact caustique (attaques des tissus des organismes aquatiques) et un autre mutagène (dommages chromosomiques) et allergisant.Les dérivés du fluorure sont abondants dans la nature. Le fluorure est utilisé dans l’industrie chimique, la production d’acide phosphorique et la production d’engrais phosphorés. Les organismes qui vivent dans les régions riches en fluor peuvent avoir des perturbations de leur comportement. Chez l’homme, le fluorure provoque la maladie de « Fluorose » et des caries dentaires et à des effets neurotoxique au niveau de la glande pinéale.